
Journal of Ocular Biology
Download PDF
Review Article
*Address for Correspondence: Mary J Kelley, Casey Eye Institute, Department of Ophthalmology, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, Oregon, USA, Tel: 5034943594; E-mail: kelleyma@ohsu.edu
Citation: Abu-Hassan DW, Acott TS, Kelley MJ. The Trabecular Meshwork: A Basic Review of Form and Function. J Ocular Biol. 2014;2(1): 9.
Copyright © 2013 Abu-Hassan DW, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 2, Issue: 1
Submission: 21 July 2014 | Accepted: 23 July 2014 | Published: 29 July 2014
Reviewed & Approved by: Dr. Eric W Hein, Assistant Professor, Department of Ophthalmology, Wake Forest Baptist Health Eye Center, USA


 The TM itself is a tiny porous triangle approximately 350 x 50-150 μm in cross section and SC is approximately 25 mm long in circumferential length [14]. The structure is composed of connective tissue beams and sheets or lamellae covered by TM cells. Three consecutive regions define the filtering portion of the TM: uveal meshwork, corneoscleral meshwork and juxtacanalicular region (JCT) also often called the cribriform region. There is also a fourth region, the non-filtering anterior part of the triangle that “inserts” under the cornea, the TM insert region (Figure 4) [28,29]. The uveal meshwork consists of approximately 3 layers of connective tissue beams covered by flat confluent TM cells which form irregular intratrabecular fenestrations [29-34]. These beams or lamellae are only found in the uveal meshwork and the next region, the corneoscleral meshwork. The latter region is composed of 8-15 layers of perforated sheets of fibers or beams, also covered by TM cells and the intratrabecular spaces become smaller as they extend closer to SC [29-34]. The cells of the outer layers of TM act primarily as pre-filters and are aggressively phagocytic, removing cellular debris from AH before the fluid moves deeper into the less porous JCT and SC. The JCT cells are more stellate and exhibit bridging contacts between the endothelial cells of SC and the TM beam cells. The JCT region appears to contain nearly all of the resistance to aqueous humor outflow. It is the thinnest part of the TM, measuring 2-20 μm thick, and consists of 2-5 amorphous and discontinuous cell layers found in a loose connective tissue ECM. This is directly adjacent to the inner wall endothelium of SC [30-34]. The JCT cells reside on a random, apparently disorganized ECM basement membrane which is attached to other ECM components [12,31,34-39]. The JCT cells attach to one another and to the endothelial cell lining of SC by long cytoplasmic processes [31-34,40-43]. The endothelial cell lining of Schlemm’s canal exhibits strong occludens or tight junctions and provides a small but significant barrier to fluid passage [38,39,44-46]. These cells form unusual drainage structures originally known as giant vacuoles (discussed later) through which AH passes to enter SC. The lumen of Schlemm’s canal is connected to the venous drainage system through an array of around 35 collector channels. Abnormal development of the various ocular drainage structures at the iridocorneal angle can lead to structural abnormalities, resulting in congenital glaucoma and an elevated IOP.
The TM itself is a tiny porous triangle approximately 350 x 50-150 μm in cross section and SC is approximately 25 mm long in circumferential length [14]. The structure is composed of connective tissue beams and sheets or lamellae covered by TM cells. Three consecutive regions define the filtering portion of the TM: uveal meshwork, corneoscleral meshwork and juxtacanalicular region (JCT) also often called the cribriform region. There is also a fourth region, the non-filtering anterior part of the triangle that “inserts” under the cornea, the TM insert region (Figure 4) [28,29]. The uveal meshwork consists of approximately 3 layers of connective tissue beams covered by flat confluent TM cells which form irregular intratrabecular fenestrations [29-34]. These beams or lamellae are only found in the uveal meshwork and the next region, the corneoscleral meshwork. The latter region is composed of 8-15 layers of perforated sheets of fibers or beams, also covered by TM cells and the intratrabecular spaces become smaller as they extend closer to SC [29-34]. The cells of the outer layers of TM act primarily as pre-filters and are aggressively phagocytic, removing cellular debris from AH before the fluid moves deeper into the less porous JCT and SC. The JCT cells are more stellate and exhibit bridging contacts between the endothelial cells of SC and the TM beam cells. The JCT region appears to contain nearly all of the resistance to aqueous humor outflow. It is the thinnest part of the TM, measuring 2-20 μm thick, and consists of 2-5 amorphous and discontinuous cell layers found in a loose connective tissue ECM. This is directly adjacent to the inner wall endothelium of SC [30-34]. The JCT cells reside on a random, apparently disorganized ECM basement membrane which is attached to other ECM components [12,31,34-39]. The JCT cells attach to one another and to the endothelial cell lining of SC by long cytoplasmic processes [31-34,40-43]. The endothelial cell lining of Schlemm’s canal exhibits strong occludens or tight junctions and provides a small but significant barrier to fluid passage [38,39,44-46]. These cells form unusual drainage structures originally known as giant vacuoles (discussed later) through which AH passes to enter SC. The lumen of Schlemm’s canal is connected to the venous drainage system through an array of around 35 collector channels. Abnormal development of the various ocular drainage structures at the iridocorneal angle can lead to structural abnormalities, resulting in congenital glaucoma and an elevated IOP.
 The ECM of the TM contains numerous structural and organizational components such as, collagens, laminins, elastin, fibronectin, fibrillins, proteoglycans, matricellular proteins, etc. [12,35,47,48]. Intratrabecular spaces of the TM allow passage of aqueous humor to SC. The spaces between JCT and ECM fibers contain a ground substance consisting of various proteoglycans and hyaluronan and serve as an outflow pathway for AH [12,30,34,49-55]. The open spaces, or fenestrations, between the lamellae are approximately 25-27 μm in the uveal meshwork and 2-15 μm in the corneoscleral meshwork, hence they form a highly porous structure and provide insignificant resistance to aqueous humor outflow. The ECM of the JCT, however, is thought to provide nearly all of the flow resistance to AH passage [12,56-62]. The center or stroma of the trabecular lamellae or beams contains densely packed collagen and elastic fibers, and the electron dense microfibril sheath-derived (SD) material [32,37,45,63-65]. The collagen structural fibers are mostly type I and III [34,66,67]. The TM cells on the beams attach to a basal lamina that contains collagen IV, laminin and a wide array of other ECM components [12,35,47,49,50,52,66,67].
The ECM of the TM contains numerous structural and organizational components such as, collagens, laminins, elastin, fibronectin, fibrillins, proteoglycans, matricellular proteins, etc. [12,35,47,48]. Intratrabecular spaces of the TM allow passage of aqueous humor to SC. The spaces between JCT and ECM fibers contain a ground substance consisting of various proteoglycans and hyaluronan and serve as an outflow pathway for AH [12,30,34,49-55]. The open spaces, or fenestrations, between the lamellae are approximately 25-27 μm in the uveal meshwork and 2-15 μm in the corneoscleral meshwork, hence they form a highly porous structure and provide insignificant resistance to aqueous humor outflow. The ECM of the JCT, however, is thought to provide nearly all of the flow resistance to AH passage [12,56-62]. The center or stroma of the trabecular lamellae or beams contains densely packed collagen and elastic fibers, and the electron dense microfibril sheath-derived (SD) material [32,37,45,63-65]. The collagen structural fibers are mostly type I and III [34,66,67]. The TM cells on the beams attach to a basal lamina that contains collagen IV, laminin and a wide array of other ECM components [12,35,47,49,50,52,66,67].
 To study IOP homeostasis in perfused anterior segment organ culture, flow is stabilized for a day or two at 1x pressure, which is around 8 mm Hg in normal eyes. Flow rates are around 2-4 μl/min, which is physiologic [10]. To trigger a homeostatic corrective response, perfusion pressure is doubled, increasing it to around 16 mm Hg. The flow rate immediately doubles but over the next day or so, the outflow resistance is correctively reduced by the TM cells attempting to restore normal IOP levels and the outflow rate gradually increases in response (Figure 6) [13]. This robust IOP homeostatic response is arguably the primary reason that most people do not ever develop glaucoma.
To study IOP homeostasis in perfused anterior segment organ culture, flow is stabilized for a day or two at 1x pressure, which is around 8 mm Hg in normal eyes. Flow rates are around 2-4 μl/min, which is physiologic [10]. To trigger a homeostatic corrective response, perfusion pressure is doubled, increasing it to around 16 mm Hg. The flow rate immediately doubles but over the next day or so, the outflow resistance is correctively reduced by the TM cells attempting to restore normal IOP levels and the outflow rate gradually increases in response (Figure 6) [13]. This robust IOP homeostatic response is arguably the primary reason that most people do not ever develop glaucoma.
 We have studied the molecular details of this process and it appears to be an ECM turnover event, which is triggered by TM and perhaps to some extent, SC cells sensing the sustained pressure change as mechanical stretching and distortion [12,13,72,77,78]. Presumably, integrins and other cell-surface ECM receptors feel the stretching and distortion of JCT ECM molecules and initiate corrective responses to adjust the outflow resistance [12,13]. Matrix metalloproteinases are activated and their translation increased to initiate the ECM turnover [12,79]. This includes complex signal transduction events and a program of ECM component expression with subsequent outflow resistance modifications [48,72,77,79-83]. Ongoing ECM turnover is required to maintain IOP and manipulations of this turnover process and its components modulate outflow [12,48,78-82,84-87].
We have studied the molecular details of this process and it appears to be an ECM turnover event, which is triggered by TM and perhaps to some extent, SC cells sensing the sustained pressure change as mechanical stretching and distortion [12,13,72,77,78]. Presumably, integrins and other cell-surface ECM receptors feel the stretching and distortion of JCT ECM molecules and initiate corrective responses to adjust the outflow resistance [12,13]. Matrix metalloproteinases are activated and their translation increased to initiate the ECM turnover [12,79]. This includes complex signal transduction events and a program of ECM component expression with subsequent outflow resistance modifications [48,72,77,79-83]. Ongoing ECM turnover is required to maintain IOP and manipulations of this turnover process and its components modulate outflow [12,48,78-82,84-87].
The Trabecular Meshwork: A Basic Review of Form and Function
Diala W Abu-Hassan1,2,3, Ted S Acott1,2 and Mary J Kelley1,4*
- 1Casey Eye Institute, Department of Ophthalmology, Oregon Health & Science University, Portland, Oregon, USA
- 2Department of Biochemistry & Molecular Biology, Oregon Health & Science University, Portland, Oregon, USA
- 3Department of Biochemistry & Physiology, University of Jordan, Amman, Jordan
- 4Department of Integrative Biosciences, Oregon Health & Science University, Portland, Oregon, USA
*Address for Correspondence: Mary J Kelley, Casey Eye Institute, Department of Ophthalmology, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, Oregon, USA, Tel: 5034943594; E-mail: kelleyma@ohsu.edu
Citation: Abu-Hassan DW, Acott TS, Kelley MJ. The Trabecular Meshwork: A Basic Review of Form and Function. J Ocular Biol. 2014;2(1): 9.
Copyright © 2013 Abu-Hassan DW, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 2, Issue: 1
Submission: 21 July 2014 | Accepted: 23 July 2014 | Published: 29 July 2014
Reviewed & Approved by: Dr. Eric W Hein, Assistant Professor, Department of Ophthalmology, Wake Forest Baptist Health Eye Center, USA
Background
Glaucoma is a group of optic neuropathies in which damage to the optic nerve results in a distinctive pattern of irreversible vision loss. Elevations in the intraocular pressure (IOP) are a common contributor to this damage. This major cause of blindness affects mainly the elderly population. Over 70 million people are diagnosed with glaucoma worldwide and over 4 million have lost vision in both eyes [1-5]. The overall prevalence of open angle glaucoma is estimated to be 3.54% [6].Glaucoma accounts for around 25% of new blindness in developed countries [3,7]. Primary open-angle glaucoma (POAG) is the most common form and constitutes up to 85% of diagnosed cases. The risk factors include elevated IOP, age, genetic factors, race, thin corneas and abnormal optic nerve anatomy [7]. Glaucoma is normally asymptomatic and is usually not detected until significant irreversible vision loss, which is primarily peripherally located in the field of view, has taken place. The therapeutic management of glaucoma costs the United States and the United Kingdom about 2.5 billion and 216 million USD annually, respectively [8,9].
IOP is a consequence of aqueous humor inflow balanced against aqueous humor outflow and both are approximately 2.75 μl/min [10]. Inflow is relatively pressure insensitive until very high pressures are achieved [11]. IOP is regulated primarily by controlled adjustments of the outflow resistance which resides in the trabecular meshwork (TM) [12,13]. Since the eye is a single hydraulic unit, pressure everywhere within the eye is uniform and IOP elevations impact the optic nerve directly, since it is most vulnerable to pressure.
Development and Structure of the TM
The anterior segment includes the cornea, lens, iris, ciliary body and ocular drainage tissues, primarily the TM and Schlemm’s canal (SC) [14]. The TM and SC are located in the iridocorneal angle, where the iris and cornea meet and the sclera transitions into the cornea [15]. The ocular drainage structures are among the last to differentiate during anterior eye development. By the 6th week of embryonic development, the rudimentary eye is composed of the bi-layered optic cup and the lens vesicle (Figure 1) [16]. The optic cup is formed from forebrain neuroectoderm while the lens vesicle invaginates and separates from the overlying surface ectoderm [16-19]. At this developmental stage, mesenchymal progenitor cells encircle the developing eye, thus they are called “periocular mesenchyme”, and these cells then migrate anteriorly [16-21]. The TM is derived from periocular mesenchyme that consists of neural crest and cranial paraxial mesoderm derived cells [15], whereas, SC is also likely derived from periocular mesenchyme, but is formed as a result of vasculature remodeling in the corneoscleral transition zone [17-19,22,23]. During human eye development, at the 15th to 20th week of gestation, the iridocorneal angle is occupied by a dense mass of mesenchymal cells shortly after iris elongation begins. In the following steps, these cells elongate, flatten, and become separated from each other by small fenestrations that are partially filled with extracellular fibers. Although the TM appears at the 15th to 20th week of gestation, the aqueous humor only begins to be secreted in the fifth to sixth month of gestation [21,24-26]. The major morphogenesis of the TM is complete around the time of birth in humans, however, in the postnatal period, significant development of the anterior segment structures, specifically, the ocular drainage structures occurs [18,27]. At this stage, the presumptive site of the TM at the iridocorneal angle contains a mass of packed mesenchymal cells (Figure 2 and 3). To allow the outflow of the aqueous humor (AH), remodeling of this mesenchymal mass and the formation of a functional TM must occur [18]. A major change required for maturation of a functional TM is the formation of intertrabecular spaces or fenestrations between a network of beams and sheets comprised of extracellular matrix (ECM) covered by TM cells.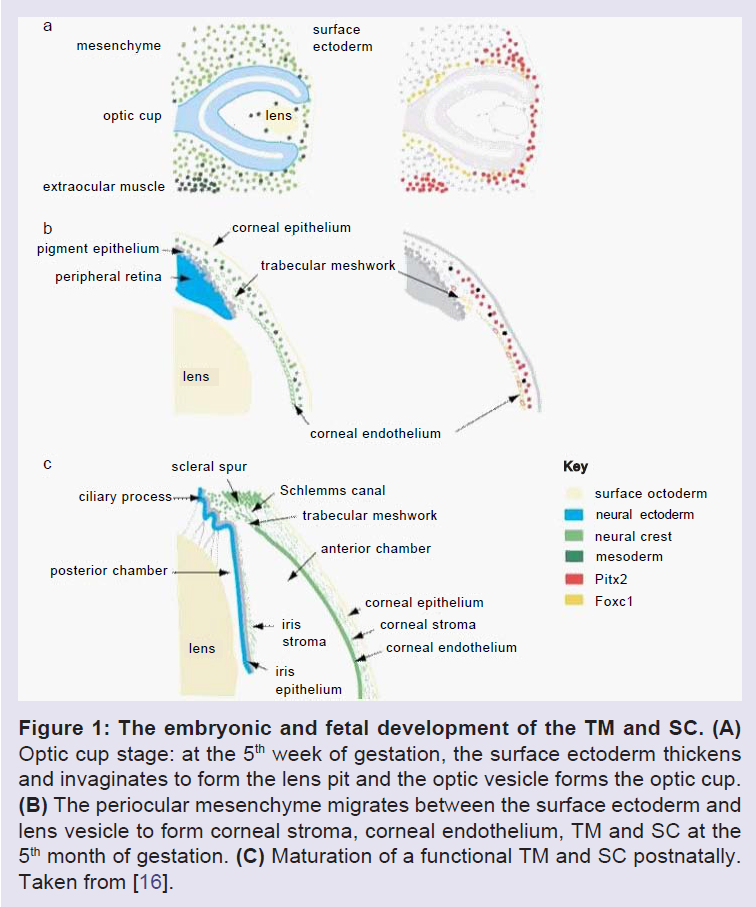
Figure 1: The embryonic and fetal development of the TM and SC. (A) Optic cup stage: at the 5th week of gestation, the surface ectoderm thickens and invaginates to form the lens pit and the optic vesicle forms the optic cup. (B) The periocular mesenchyme migrates between the surface ectoderm and lens vesicle to form corneal stroma, corneal endothelium, TM and SC at the 5th month of gestation. (C) Maturation of a functional TM and SC postnatally. Taken from [16].
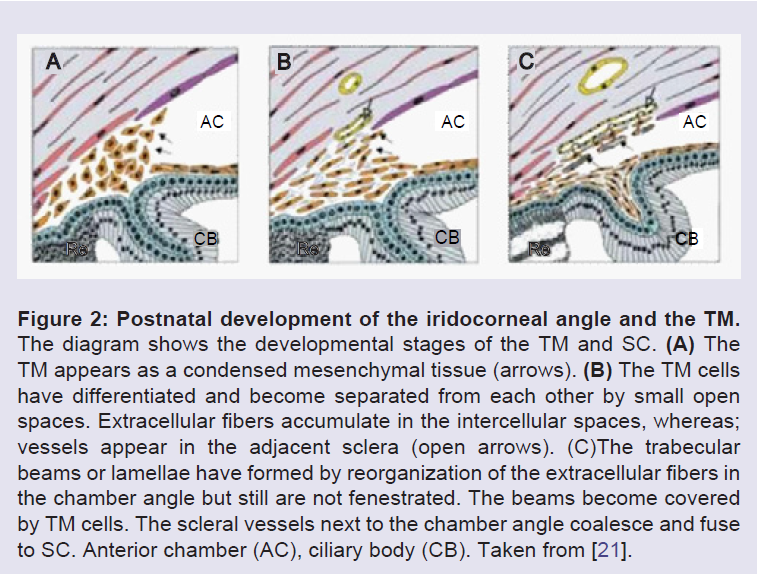
Figure 2: Postnatal development of the iridocorneal angle and the TM. The diagram shows the developmental stages of the TM and SC. (A) The TM appears as a condensed mesenchymal tissue (arrows). (B) The TM cells have differentiated and become separated from each other by small open spaces. Extracellular fibers accumulate in the intercellular spaces, whereas; vessels appear in the adjacent sclera (open arrows). (C) The trabecular beams or lamellae have formed by reorganization of the extracellular fibers in the chamber angle but still are not fenestrated. The beams become covered by TM cells. The scleral vessels next to the chamber angle coalesce and fuse to SC. Anterior chamber (AC), ciliary body (CB). Taken from [21].
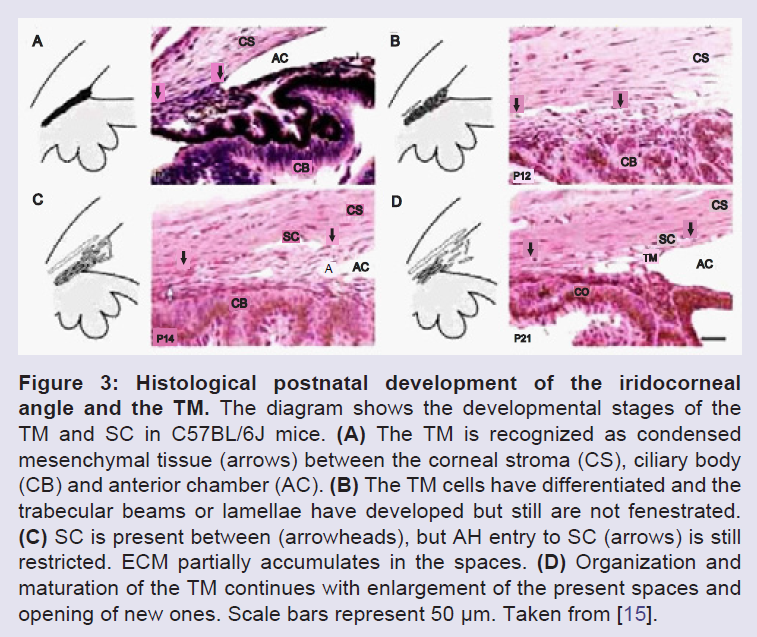
Figure 3: Histological postnatal development of the iridocorneal angle and the TM. The diagram shows the developmental stages of the TM and SC in C57BL/6J mice. (A) The TM is recognized as condensed mesenchymal tissue (arrows) between the corneal stroma (CS), ciliary body (CB) and anterior chamber (AC). (B) The TM cells have differentiated and the trabecular beams or lamellae have developed but still are not fenestrated. (C) SC is present between (arrowheads), but AH entry to SC (arrows) is still restricted. ECM partially accumulates in the spaces. (D) Organization and maturation of the TM continues with enlargement of the present spaces and opening of new ones. Scale bars represent 50 μm. Taken from [15].
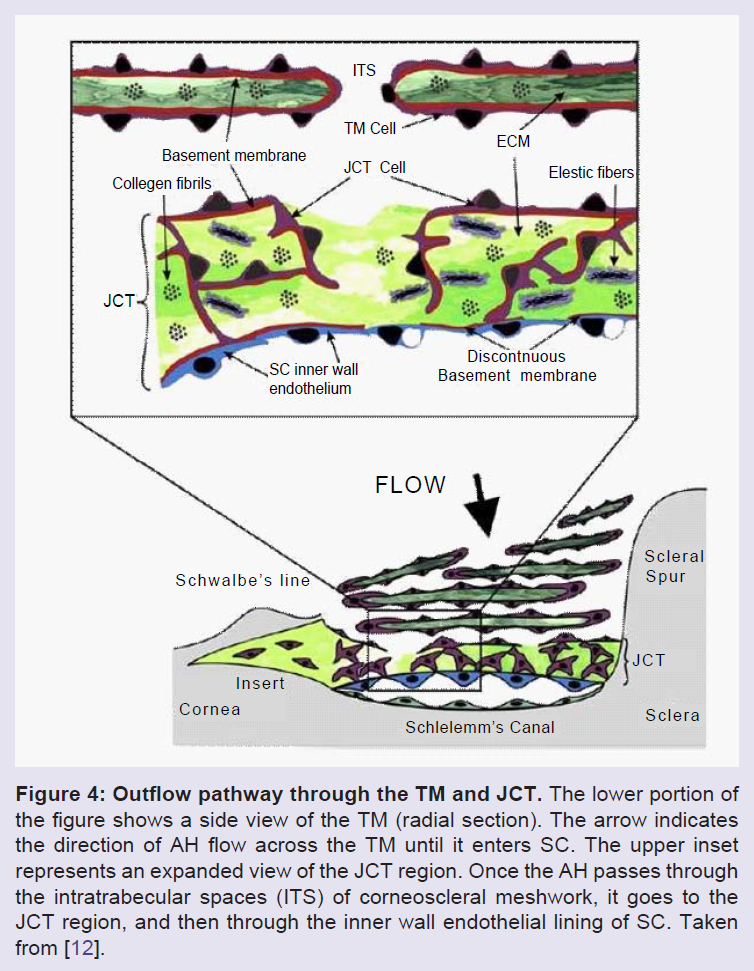
Figure 4: Outflow pathway through the TM and JCT. The lower portion of the figure shows a side view of the TM (radial section). The arrow indicates the direction of AH flow across the TM until it enters SC. The upper inset represents an expanded view of the JCT region. Once the AH passes through the intratrabecular spaces (ITS) of corneoscleral meshwork, it goes to the JCT region, and then through the inner wall endothelial lining of SC. Taken from [12].
The Aqueous Pathway, TM Physiology, and IOP Maintenance
AH fills the anterior and posterior chambers of the eye in front of the lens and is produced in the ciliary body within the posterior chamber. After filling the posterior chamber, AH moves anteriorly across the lens and around the iris then passes through the pupil to the anterior chamber. It nourishes the avascular lens, cornea and TM and inflates the eye to provide sharp focused vision [29,64,68]. The AH contains cellular debris from upstream tissues that is removed by the outer layer of TM cells, which are aggressively phagocytic. Most of the AH exits via the conventional, TM/SC route. Around 10% leaves via the unconventional or uveoscleral pathway through the intercellular spaces of the ciliary muscle cells and this ratio may change in glaucoma [29,37,69-71].Acute fluctuations in IOP occur frequently as a result of many factors including daily variation, exercise, heart rate, respiration, fluid intake, medications, body orientation and alcohol consumption. In addition, IOP can drift because of longer term outflow resistance changes. In response to sustained imbalances in IOP, TM cells can mount a homeostatic response to adjust the outflow resistance and restore the IOP level to within normal narrow physiological ranges [12,13,72]. The most common experimental method to study IOP regulation is an ex vivo system called perfused anterior segment organ culture (Figure 5) [73-76]. Studies can be conducted at constant flow, i.e. stable peristaltic pumping, with IOP measurement via pressure transducers, or at constant pressure with analysis of flow rates, usually gravimetrically. Studies with human tissue are normally feasible for use for 2-3 weeks. Perhaps the most important feature of the perfusion system is this ability to mimic the normal IOP homeostatic effect, in which sustained pressure deviations are sensed and the TM responds by adjusting the outflow resistance slowly over a day or two’s time [12,13,72]. IOP is then returned to within the narrow bounds that are normally observed in vivo. Although several excellent animal models, particularly the mouse, are often used for IOP studies, currently there is not an easy unbiased method to study this central IOP homeostatic response directly [13]. These models are quite useful in evaluating the effects of gene manipulations, drugs, and treatments on outflow and IOP and many of these studies are likely impacting components of the normal IOP homeostatic process.
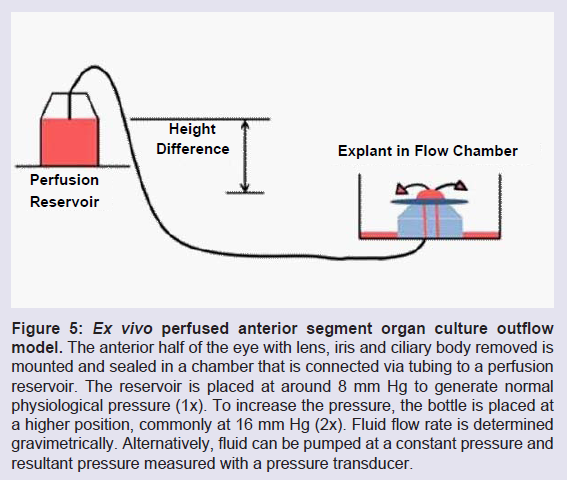
Figure 5: Ex vivo perfused anterior segment organ culture outflow model. The anterior half of the eye with lens, iris and ciliary body removed is mounted and sealed in a chamber that is connected via tubing to a perfusion reservoir. The reservoir is placed at around 8 mm Hg to generate normal physiological pressure (1x). To increase the pressure, the bottle is placed at a higher position, commonly at 16 mm Hg (2x). Fluid flow rate is determined gravimetrically. Alternatively, fluid can be pumped at a constant pressure and resultant pressure measured with a pressure transducer.
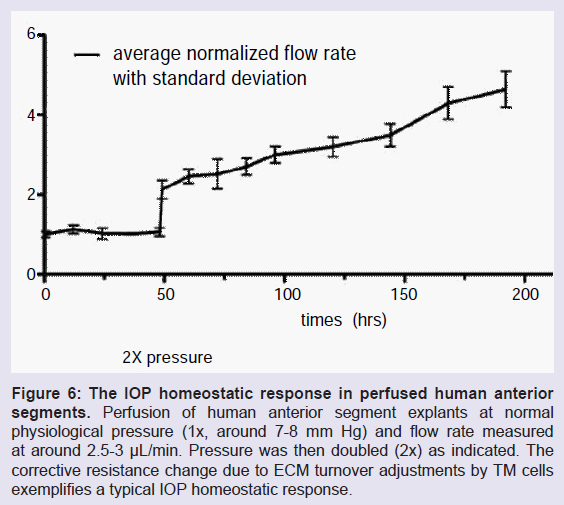
Figure 6: The IOP homeostatic response in perfused human anterior segments. Perfusion of human anterior segment explants at normal physiological pressure (1x, around 7-8 mm Hg) and flow rate measured at around 2.5-3 μL/min. Pressure was then doubled (2x) as indicated. The corrective resistance change due to ECM turnover adjustments by TM cells exemplifies a typical IOP homeostatic response.
Since the outflow resistance is thought to be primarily comprised of ECM components, perhaps predominantly versican, this process is particularly relevant [12,79,81,88]. Most studies have concluded that all but a small fraction, probably less than 10%, of the outflow resistance resides in the ECM of the deepest portion of the JCT and inner wall SC basement membrane [12,56-58,89,90]. With the effect of “funneling” the SC inner wall cells could restrict fluid egress to only a few active pores at a time, i.e. the neck of funnel [56,58,89,90]. Instead of the whole JCT ECM being the site of fluid passage, only a small portion of it would be actively passing fluid. This restriction would force AH to flow through only a small portion of the ECM near the inner wall at or near the sites of the pores. Although SC cells clearly could not sustain the full pressure drop across the TM (around 7-9 mm Hg), by the time AH reached the inner wall most of the pressure would be dissipated by passing through the flow resistance area of the ECM of the JCT.
The giant vacuoles and pores which form where AH passes through SC inner wall endothelial cells have been intriguing to many for years. They are thought to be the site of egress of AH through this very tight endothelial cell monolayer. In response to the pressure gradient generated by AH flow, the SC inner wall endothelium forms cellular outpouchings, the giant vacuoles. These characteristic structures form when AH pushes against the basal side of SC cells [39,69,70,91-93]. Inner wall cells of SC rest on an incomplete basal lamina and are not supported by ECM in some areas of their basal cell membrane [34]. The AH passes through SC via both intracellular (I-pores) and paracellular or border pores (B-pores) [89,94-97]. I-pores have diameters from 0.1 to 2 μm and are often associated with giant vacuoles [94]. Although B-pores usually have similar diameters, they are less common in most eyes [94]. The endothelial cells that line SC are held together by tight occludens junctions [98]. Particles 200-500 nm in size can cross the inner wall pores [98]. Although the inner wall endothelium provides some hindrance to AH outflow, as mentioned above it appears not to generate more than 10% of the total outflow resistance [99]. The molecular mechanisms that cause the formation of intracellular pores in SC inner wall endothelium are still unknown. Additionally, small diaphragmed minipores were identified in the inner wall of SC [30,91]. These minipores may represent an early stage in pore formation and are ultrastructurally similar to those forming the fenestrae of fenestrated capillaries [30,100]. They appear to involve caveolin proteins, which have been associated with glaucoma and which appear to serve some key roles in outflow [101,102]. Additional studies of these pores may clarify their role and contributions to the outflow resistance and outflow regulation [57,89,90,94,103-105].
Further complicating the outflow picture is the observation that aqueous outflow is highly segmental in nature [63,81,105-111]. This means that only a portion, perhaps a third, of the outflow pathway is active in fluid passage at any one time. The molecular differences between high and low flow regions will thus be particularly pertinent. Distribution patterns of ECM and regulatory molecules between these high and low flow regions currently are being determined. SPARC knockdown was recently shown to increase the proportion of high flow areas relative to low flow areas in mice [112]. Understanding regulation of the high and low flow distribution promises to be highly informative in the immediate future.
TM cells change the expression levels of numerous ECM components in response to mechanical stretching or pressure, TNFα, IL-1, TGFβ2 and dexamethasone. All are agents that manipulate outflow resistance [12]. The expression of tenascin, fibronectin, CD44, syndecan 2, SPARC, VCAM-1, Collagen XIV, Periostin, Matrix Gla protein, Link protein and MMP-2 is increased with stretching, whereas, versican, syndecan 1, and thrombospondin 2 are reduced [48]. Coincident with these changes over the next few days, the flow rate increase as the outflow resistance is reduced [72]. ECM turnover is a highly coordinated sequence of events that involves the degradation, biosynthetic replacement and altered organization of specific ECM components [12].
Recently, a mechanism for controlled focal ECM turnover involving podosome- or invadopodia-like structures (PILS) containing ECM adhesion and degradation molecules has been identified at the site of TM ECM turnover [113]. This process includes MMP2 and MMP14 with co-localization of a number of cytoskeletal and vesicle coordination molecules [113]. These highly-regulated focal PILS conduct ECM degradation and uptake, ECM recycling, and may be involved in laying down replacement ECM during remodeling of the outflow resistance.
Glaucoma
In glaucoma, the flow of AH is often obstructed resulting in pressure elevation in the eye, since inflow remains unchanged. The increased ocular pressure causes degenerative changes and progressive death of the retinal ganglion cells (RGCs) and damages their axons, which transfer visual information to the brain via the optic nerve [64,68]. Increased IOP, which is a risk factor for POAG, is directly related to increases in outflow resistance [114,115]. The difficulty of the passage of AH through the TM, represented by flow resistance, has been attributed mainly to the innermost region of the TM, the JCT [12,58]. A wide variety of changes in the organization, structure and integrity of the TM tissue have been detected in glaucomatous eyes. The most characteristic structural changes are the accumulation of ECM and banded fibrillar elements that are embedded in different glycoproteins, known as “plaque material” [34,36,116-123]. The role of these and other changes with glaucoma are not clearly established at this time. Recently, strong relationships between ECM stiffness and glaucoma have been reported. One study found glaucoma JCT ECM approximately 20-fold stiffer while another study found the opposite [124,125]. Since the methodology was very different, additional studies will be required to resolve the implications of these observations.Additionally, in glaucoma TM cell counts decrease with aging and even more with POAG [126-128]. The JCT specifically shows more cell loss than other layers of the TM, nevertheless, the total cell number in the other layers of the TM is reduced as well. Mechanical stresses, oxidative damage, time, disease or other injuries also may contribute to cell loss or dysfunction in the TM. The cause(s) of the increased cell loss is/are not yet established, but this observation does have implications for glaucoma in general. TM cellularity is increased following laser trabeculoplasty, a common glaucoma treatment, with TM cell division increased by 4-fold following laser-treatment over non-treated controls [28,129,130]. More than 60% of the cell division was detected in the anterior, non-filtering region of the TM, or the insert area. In this area, the TM inserts into the cornea beneath Schwalbe’s line, and “stem-like cells”, called TM insert cells, are present. A week after laser trabeculoplasty, 60% of tritiated thymidinelabeled cells migrated out of the insert region to repopulate laser burn sites [28]. This study suggests that the insert cells serve as TM stem cells and that the repopulation of the TM tissue by fresh, healthy, and functioning cells could restore the role the tissue performs in keeping eye pressure normal [131].
Molecular and Biochemical Mechanisms of Glaucoma
Changes in the normal structure of the TM also have been detected with aging and in POAG. An increase in the thickness of the ECM in the JCT region has been observed in glaucomatous eyes compared to healthy controls [37]. Other investigations suggested the formation of deposits by the interacting ECM components, thus blocking the outflow pathway through TM. For instance, cochlin was identified by a proteomic analysis to be associated with one type of glaucomatous TM but was not in normal TM. Induced by shear stress, cochlin undergoes multimerization and forms deposits that have exclusively been found with mucopolysaccharide deposits in glaucomatous TM [132-135]. Other changes apparent with glaucoma were trabecular thickening and fusions, hyperpigmentation of TM cells, and the loss of giant vacuoles from the inner wall of Schlemm’s canal. Moreover, there is a significant increase in the amount of sheath derived (SD)-plaques in glaucomatous compared to normal eyes [136]. Additional fibrillar material that adheres to the net of the TM results in an irregular thickness of the sheath. In POAG eyes, increased amounts of type VI collagen surround the connecting fibrils that accumulate in advanced disease to participate in blocking the outflow pathway [36,122,123]. When the glycosaminoglycan profiles were compared between normal and glaucoma patients, chondroitin sulfate was found to be increased and hyaluronan was decreased in the JCT of glaucoma patients [137,138]. In addition, the hyaluronan receptor, CD44, exhibits complex possible relationships to glaucoma [139-143]. Although the specific interpretation is not immediately obvious, this likely has major implications for the glaucoma mechanism. There are a large number of other intriguing observations that are yet to be fully understood as they relate to glaucoma. Most of the exact molecular mechanisms that cause glaucoma neither are needed still unclear [65].The contractility of the TM also affects aqueous drainage where increased contractility reduces AH outflow and relaxation increases AH drainage [144-148]. Therefore, the disruption of the cytoskeleton greatly disturbs the structural integrity and functionality of the TM tissue. Among the structural changes that were observed in glaucomatous TM is the formation of cross-linked actin networks (CLANs). These polygonal structures are thought to increase the rigidity and stiffness of the TM cells rendering them more resistant to aqueous outflow [149-157].
Since the JCT is where most of the flow resistance is located, ECM remodeling is of special importance in this particular region. Both autocrine and paracrine growth factors, such as TGF-β2, CTGF, and bone morphogenic proteins (BMP7 and BMP4), control the synthesis and degradation of ECM molecules in the JCT [158-162]. Moreover, glucocorticoids and prostaglandin derivatives modulate ECM turnover in the JCT [163-169]. An increase in fibrillar content in the ECM of the JCT is a prominent structural change noted in eyes of patients with POAG and steroid-induced glaucoma [170]. ECM related herniations of the JCT and the inner wall of SC have been identified but their molecular structure is still unknown [109]. Other studies have also detected an induction of calcification markers such as matrix Gla protein and BMP-2 [171-174], thereby suggesting that calcification of the TM may contribute to the stiffness and increased flow resistance of glaucomatous tissue [124].
In summary, glaucoma and the involvement of the outflow pathway in normal and pathological processes remains only partially understood. Further investigations with models, with cell and organ culture, with molecular mechanisms, and with pharmaceutical options may eventually unravel and elucidate this complex blinding disease.
Acknowledgements
This work was supported by NIH grants EY021800 (MJK), EY003279 (TSA), EY008247 (TSA), and EY010572 (TSA), an unrestricted grant to the Casey Eye Institute from Research to Prevent Blindness, New York, New York, and by the University of Jordan (DWA).References
- Quigley HA (1993) Open-angle glaucoma. N Eng J Med 328: 1097-1106.
- Quigley HA (1996) Number of people with glaucoma worldwide. Br J Ophthalmol 80: 389-393.
- Quigley HA, Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90: 262-267.
- Quigley HA, Vitale S (1997) Models of open-angle glaucoma prevalence and incidence in the United States. Invest Ophthalmol Vis Sci 38: 83-91.
- Thylefors B, Negrel AD (1994) The global impact of glaucoma. Bull World Health Organ 72: 323-326.
- Tham YC, Li X, Wong TY, Quigley HA, Aung T, et al. (2014) Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology [Epub ahead of print].
- Quigley HA (2011) Glaucoma. Lancet 377: 1367-1377.
- Traverso CE, Walt JG, Kelly SP, Hommer AH, Bron AM, et al. (2005) Direct costs of glaucoma and severity of the disease: a multinational long term study of resource utilisation in Europe. Br J Ophthalmol 89: 1245-1249.
- Kobelt-Nguyen G, Gerdtham UG, Alm A (1998) Costs of treating primary open-angle glaucoma and ocular hypertension: a retrospective, observational two-year chart review of newly diagnosed patients in Sweden and the United States. J Glaucoma 7: 95-104.
- Brubaker R (1996) Measurement of aqueous flow by fluorophotometry., in The Glaucomas, Ritch R, Shields, MB, Krupin T, Editor, Mosby: St Louis. p. 447-454.
- Brubaker RF (1970) The measurement of pseudofacility and true facility by constant pressure perfusion in the normal rhesus monkey eye. Invest Ophthalmol 9: 42-52.
- Acott TS, Kelley MJ (2008) Extracellular matrix in the trabecular meshwork. Exp Eye Res 86: 543-561.
- Acott TS, Kelley MJ, Keller KE, Vranka JA, Abu-Hassan DW, et al. (2014) Intraocular pressure homeostasis: maintaining balance in a high-pressure environment. J Ocul Pharmacol Ther 30: 94-101.
- Hogan MJ, Alvarado JA, Weddell JE (1971) Histology of the human eye: an atlas and textbook. Philadelphia: W. B. Saunders Company.
- Gould DB, Smith RS, John SW (2004) Anterior segment development relevant to glaucoma. Int J Dev Biol 48: 1015-1029.
- Sowden JC (2007) Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 21: 1310-1318.
- Smith RS, Zabaleta A, Savinova OV, John SW (2001) The mouse anterior chamber angle and trabecular meshwork develop without cell death. BMC Dev Biol 1: 3.
- Johnston MC, Noden DM, Hazelton RD, Coulombre JL, Coulombre AJ (1979) Origins of avian ocular and periocular tissues. Exp Eye Res 29: 27-43.
- Trainor PA, Tam PP (1995) Cranial paraxial mesoderm and neural crest cells of the mouse embryo: co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development 121: 2569-2582.
- Reme C, d’Epinay SL (1981) Periods of development of the normal human chamber angle. Doc Ophthalmol 51: 241-268.
- Cvekl A, Tamm ER (2004) Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays 26: 374-386.
- 22. Hamanaka T, Bill A, Ichinohasama R, Ishida T (1992) Aspects of the development of Schlemm’s canal. Exp Eye Res 55: 479-488.
- Noden DM (1975) An analysis of migratory behavior of avian cephalic neural crest cells. Dev Biol 42: 106-130.
- Wulle KG (1968) Electron microscopic observations of the development of Schlemm’s canal in the human eye. Trans Am Acad Ophthalmol Otolaryngol 72: 765-773.
- Wulle KG (1972) Electron microscopy of the fetal development of the corneal endothelium and Descemet’s membrane of the human eye. Invest Ophthalmol 11: 897-904.
- Mao M, Hedberg-Buenz A, Koehn D, John SW, Anderson MG (2011) Anterior segment dysgenesis and early-onset glaucoma in nee mice with mutation of Sh3pxd2b. Invest Ophthalmol Vis Sci 52: 2679-2688.
- Baulmann DC, Ohlmann A, Flugel-Koch C, Goswami S, Cvekl A, et al. (2002) Pax6 heterozygous eyes show defects in chamber angle differentiation that are associated with a wide spectrum of other anterior eye segment abnormalities. Mech Dev 118: 3-17.
- Acott TS, Samples JR, Bradley JM, Bacon DR, Bylsma SS, et al. (1989) Trabecular repopulation by anterior trabecular meshwork cells after laser trabeculoplasty. Am J Ophthalmol 107: 1-6.
- Shields MB (1998) Textbook of Glaucoma. 4th ed., Baltimore: Williams & Wilkins.
- Tamm ER (2009) The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res 88: 648-655.
- Lutjen-Drecoll E (1999) Functional morphology of the trabecular meshwork in primate eyes. Prog Retin Eye Res 18: 91-119.
- Lutjen-Drecoll E, Futa R, Rohen JW (1981) Ultrahistochemical studies on tangential sections of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci 21: 563-573.
- Lutjen-Drecoll E, Rohen JW (1994) Anatomy of aqueous humor formation and drainage., in Textbook of Ophthalmology, Podos SM and Yanoff M, Editors. Mosby: London. p. 1.1-1.16.
- Lutjen-Drecoll E, Rohen JW (2001) Functional Morphology of the Trabecular Meshwork., in Duane's Foundations of Clinical Ophthalmology, W. Tasman and E.A. Jaeger, Editors. J.B. Lippincott Company: Philadelphia. p. 1-30.
- Fuchshofer R, Welge-Lussen U, Lutjen-Drecoll E, Birke M (2006) Biochemical and morphological analysis of basement membrane component expression in corneoscleral and cribriform human trabecular meshwork cells. Invest Ophthalmol Vis Sci 47: 794-801.
- Lutjen-Drecoll E (2005) Morphological changes in glaucomatous eyes and the role of TGFbeta2 for the pathogenesis of the disease. Exp Eye Res 81: 1-4.
- Lutjen-Drecoll E, Rohen JW (1996) Morphology of aqueous outflow pathways in normal and glaucomatous eyes., in The Glaucomas, R. Ritch, M.B. Shields, and T. Krupin, Editors. Mosby-Year Book, Inc.: St. Louis. p. 89-124.
- Gong H, Overby D, Ruberti J, Freddo T, Johnson M (2001) Human outflow pathway viewed by quick freeze deep etch. Invest Ophthalmol Vis Sci 42: S749.
- Gong H, Tripathi RC, Tripathi BJ (1996) Morphology of the aqueous outflow pathway. Microsc Res Tech 33: 336-367.
- Grierson I, Lee WR (1974) Junctions between the cells of the trabecular meshwork. Albrecht Von Graefes Arch Klin Exp Ophthalmol 192: 89-104.
- Grierson I, Lee WR (1975) The fine structure of the trabecular meshwork at graded levels of intraocular pressure. (1) Pressure effects within the near-physiological range (8-30 mmHg). Exp Eye Res 20: 505-521.
- Grierson I, Lee WR, Abraham S, Howes RC (1978) Associations between the cells of the walls of Schlemm’s canal. Albrecht Von Graefes Arch Klin Exp Ophthalmol 208: 33-47.
- Grierson I, Lee WR, Moseley H, Abraham S (1979) The trabecular wall of Schlemm’s canal: a study of the effects of pilocarpine by scanning electron microscopy. Br J Ophthalmol 63: 9-16.
- Gong H, Brown K, Johnson M, Kamm RD, Freddo TF (1997) Hydraulic conductivity of juxtacanalicular connective tissue using quick-freeze/deep-ETCH. Invest Ophthalmol Vis Sci 38: S564.
- Gong H, Ruberti J, Overby D, Johnson M, Freddo TF (2002) A new view of the human trabecular meshwork using quick-freeze, deep-etch electron microscopy. Exp Eye Res 75: 347-358.
- Gong H, Trinkaus-Randall V, Freddo TF (1989) Ultrastructural immunoctochemical localization of elastin in normal human trabecular meshwork. Curr Eye Res 8: 1071-1082.
- Yue BY (1996) The extracellular matrix and its modulation in the trabecular meshwork. Surv Ophthalmol 40: 379-390.
- Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS (2005) Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci 46: 2857-2868.
- Acott TS (1992) Trabecular extracellular matrix regulation, in Pharmacology of Glaucoma, Drance SM, Van Buskirk EM and Neufeld AH, Editors. Williams & Wilkins: Baltimore. p. 125-157.
- Acott TS (1994) Biochemistry of aqueous humor outflow, in Textbook of Ophthalmology, Kaufman PL and Mittag TW, Editors. Mosby: London. p. 1.47-1.78.
- Acott TS (1994) Receptor biology and glaucoma - Integrins in the eye, in Encounters in glaucoma research 1: Receptor biology and glaucoma, Anderson DR and Drance SM, Editors. Foglizza Editore: Milano. 97-135.
- Acott TS, Wirtz WK (1996) Biochemistry of aqueous outflow, in The Glaucomas, Ritch R, Shields MB, and Krupin T, Editors. Mosby: St Louis. p. 281-305.
- Tawara A, Inomata H (1994) Distribution and characterization of sulfated proteoglycans in the trabecular tissue of goniodysgenetic glaucoma. Am J Ophthalmol 117: 741-755.
- Lutjen-Drecoll E, Schenholm M, Tamm E, Tengblad A (1990) Visualization of hyaluronic acid in the anterior segment of rabbit and monkey eyes. Exp Eye Research 51: 55-63.
- Gong H, Freddo TF, Johnson M (1992) Age-related changes of sulfated proteoglycans in the normal human trabecular meshwork. Exp Eye Res 55: 691-709.
- Ethier CR, Kamm RD, Palaszewski BA, Johnson MC, Richardson TM (1986) Calculations of flow resistance in the juxtacanalicular meshwork. Invest Ophthalmol Vis Sci 27: 1741-1750.
- Ethier CR, Read AT, Chan D (2004) Biomechanics of Schlemm’s canal endothelial cells: influence on F-actin architecture. Biophys J 87: 2828-2837.
- Johnson M (2006) ‘What controls aqueous humour outflow resistance?’. Exp Eye Res 82: 545-557.
- Fine BS (1966) Structure of the trabecular meshwork and the canal of Schlemm. Trans Am Acad Ophthalmol Otolaryngol 70: 777-790.
- Johnson M, Erickson K (2000) Mechanisms and routes of aqueous humor drainage., in Principles and Practice of Ophthalmology, Albert D and Jakobiec E, Editors. W.B. Saunders: Philadelphia. p. 2577-2595.
- Grant WM (1963) Experimental aqueous perfusion in enucleated human eyes. Arch Ophthalmol 69: 783-801.
- McEwen WK (1958) Application of Poiseuille’s law to aqueous outflow. AMA Arch Ophthalmol 60: 290-294.
- Buller C, Johnson D (1994) Segmental variability of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci 35: 3841-3851.
- Morrison JC, Acott TS (2003) Anatomy and Physiology of Aqueous Humor Outflow, in Glaucoma: Science and Practice, J.C. Morrison and I.P. Pollack, Editors. Thieme: New York. p. 34-41.
- Stamer WD, Acott TS (2012) Current understanding of conventional outflow dysfunction in glaucoma. Curr Opin Ophthalmol 23: 135-143.
- Marshall GE, Konstas AG, Lee WR (1990) Immunogold localization of type IV collagen and laminin in the aging human outflow system. Exp Eye Res 51: 691-699.
- Marshall GE, Konstas AG, Lee WR (1991) Immunogold ultrastructural localization of collagens in the aged human outflow system. Ophthalmology 98: 692-700.
- Morrison JC, Pollack IP (2003) Glaucoma: Science and Practice. Thieme Medical Publishers: New York, Stuttgart.
- Tripathi RC (1971) Mechanism of the aqueous outflow across the trabecular wall of Schlemm’s canal. Exp Eye Res 11: 116-121.
- Tripathi RC (1977) The functional morphology of the outflow systems of ocular and cerebrospinal fluids. Exp Eye Res 25: 65-116.
- Weinreb RN (2000) Uveoscleral outflow: the other outflow pathway. J Glaucoma 9: 343-345.
- Bradley JM, Kelley MJ, Zhu X, Anderssohn AM, Alexander JP, et al. (2001) Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci 42: 1505-1513.
- Johnson DH (1996) Human trabecular meshwork cell survival is dependent on perfusion rate. Invest Ophthalmol Vis Sci 37: 1204-1208.
- Johnson DH, Tschumper RC (1987) Human trabecular meshwork organ culture. Invest Ophthalmol Vis Sci 28: 945-953.
- Johnson DH, Tschumper RC (1989) The effect of organ culture on human trabecular meshwork. Exp Eye Res 49: 113-127.
- Tschumper RC, Johnson DH, Bradley JM, Acott TS (1990) Glycosaminoglycans of human trabecular meshwork in perfusion organ culture. Curr Eye Res 9: 363-369.
- Bradley JM, Kelley MJ, Rose A, Acott TS (2003) Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Invest Ophthalmol Vis Sci 44: 5174-5181.
- Bradley JM, Vranka J, Colvis CM, Conger DM, Alexander JP, et al. (1998) Effects of matrix metalloproteinase activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci 39: 2649-2658.
- Keller K, Aga M, Bradley JM, Kelley MJ, Acott TS (2009) Extracellular matrix turnover and outflow resistance. Exp Eye Res 88: 676-682.
- Keller K, Bradley JM, Acott TS (2009) Differential effects of ADAMTSs -1, -4, and -5 in the Trabecular Meshwork. Invest Ophthalmol Vis Sci 50: 5769-5777.
- Keller KE, Bradley JM, Vranka JA, Acott TS (2011) Segmental versican expression in the trabecular meshwork and involvement in outflow facility. Invest Ophthalmol Vis Sci 52: 5049-5057.
- Keller KE, Kelley MJ, Acott TS (2007) Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci 48: 1164-1172.
- Bradley JMB, Conger DM, Fisk AS, Samples JR, Acott TS (1995) Matrix metalloproteinases increase outflow facility in perfused anterior segment explants. Invest Ophthalmol Vis Sci 36: S727.
- Keller KE, Bradley JM, Kelley MJ, Acott TS (2008) Effects of modifiers of glycosaminoglycan biosynthesis on outflow facillity in perfusion culture. Invest Ophthalmol Vis Sci 49: 2495-2505.
- Keller KE, Sun YY, Vranka JA, Hayashi L, Acott TS (2012) Inhibition of hyaluronan synthesis reduces versican and fibronectin levels in trabecular meshwork cells. PLoS One 7: e48523.
- Keller KE, Sun YY, Vranka JA, Hayashi L, Acott TS (2012) Perturbation of hyaluronan synthesis in the trabecular meshwork and the effects on outflow facility. Invest Ophthalmol Vis Sci 53: 4616-4625.
- Bradley JM, Anderssohn AM, Colvis CM, Parshley DE, Zhu XH, et al. (2000) Mediation of laser trabeculoplasty-induced matrix metalloproteinase expression by IL-1beta and TNFalpha. Invest Ophthalmol Vis Sci 41: 422-430.
- Keller KE, Acott TS (2013) The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J Ocul Biol 1: 3.
- Ethier CR (2002) The inner wall of Schlemm’s canal. Exp Eye Res 74: 161-172.
- Overby DR, Stamer WD, Johnson M (2009) The changing paradigm of outflow resistance generation: towards synergistic models of the JCT and inner wall endothelium. Exp Eye Res 88: 656-670.
- Inomata H, Bill A, Smelser GK (1972) Aqueous humor pathways through the trabecular meshwork and into Schlemm’s canal in the cynomolgus monkey (Macaca irus): An electron microscopic study. Am J Ophthalmol 73: 760-789.
- Bill A, Maepea O (1994) Mechanisms and routes of aqueous humor drainage. in Principles and Practices of Ophthalmology, Albert DM and Jakotiec FA, Editors. W B Saunders: Philadelphia. p. 206-226.
- Johnstone MA, Grant WG (1973) Pressure-dependent changes in structures of the aqueous outflow system of human and monkey eyes. Am J Ophthalmol 75: 365-383.
- Ethier CR, Coloma FM, Sit AJ, Johnson M (1998) Two pore types in the inner-wall endothelium of Schlemm’s canal. Invest Ophthalmol Vis Sci 39: 2041-2048.
- Allingham RR, de Kater AW, Ethier CR, Anderson PJ, Hertzmark E, et al. (1992) The relationship between pore density and outflow facility in human eyes. Invest Ophthalmol Vis Sci 33: 1661-1669.
- Alvarado JA, Alvarado RG, Yeh RF, Franse-Carman L, Marcellino GR, et al. (2005) A new insight into the cellular regulation of aqueous outflow: how trabecular meshwork endothelial cells drive a mechanism that regulates the permeability of Schlemm’s canal endothelial cells. Br J Ophthalmol 89: 1500-1505.
- Alvarado JA, Betanzos A, Franse-Carman L, Chen J, Gonzalez-Mariscal L (2004) Endothelia of Schlemm’s canal and trabecular meshwork: distinct molecular, functional, and anatomic features. Am J Physiol Cell Physiol 286: C621-634.
- Ye W, Gong H, Sit A, Johnson M, Freddo TF (1997) Interendothelial junctions in normal human Schlemm’s canal respond to changes in pressure. Invest Ophthalmol Vis Sci 38: 2460-2468.
- Bill A, Svedbergh B (1972) Scanning electronmicroscopic studies of the trabecular meshwork and the canal of Schlemm: An attempt to localize the main resistance to ouflow of aqueous humor in man. Acta Ophthalmol (Copenh) 50: 295-320.
- Tamm ER, Lutjen-Drecoll E (1996) Ciliary body. Microsc Res Tech 33: 390-439.
- Loomis SJ, Kang JH, Weinreb RN, Yaspan BL, Cooke Bailey JN, et al. (2014) Association of CAV1/CAV2 genomic variants with primary open-angle glaucoma overall and by gender and pattern of visual field loss. Ophthalmology 121: 508-516.
- Wiggs JL, Kang JH, Yaspan BL, Mirel DB, Laurie C, et al. (2011) Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma in Caucasians from the USA. Hum Mol Genet 20: 4707-4713.
- Epstein D, Anderson P (1984) The Biochemistry of Outflow Mechanisms, in Glaucoma: Applied Pharmacology in Medical Treatment, Drance S, Editor. Grune & Stratton: New York. p. 135-150.
- Epstein DL, Rohen JW (1991) Morphology of the trabecular meshwork and inner-wal endothelium after cationized ferritin perfusioin in the monkey eye. Invest Ophthalmol Visl Sci 32: 160-171.
- Overby D, Gong H, Qiu G, Freddo TF, Johnson M (2002) The mechanism of increasing outflow facility during washout in the bovine eye. Invest Ophthalmol Vis Sci 43: 3455-3464.
- Chang JY, Folz SJ, Laryea SN, Overby DR (2014) Multi-scale analysis of segmental outflow patterns in human trabecular meshwork with changing intraocular pressure. J Ocul Pharmacol Ther 30: 213-223.
- Hann CR, Bahler CK, Johnson DH (2005) Cationic ferritin and segmental flow through the trabecular meshwork. Invest Ophthalmol Vis Sci 46: 1-7.
- Hann CR, Fautsch MP (2009) Preferential fluid flow in the human trabecular meshwork near collector channels. Invest Ophthalmol Vis Sci 50: 1692-1697.
- Battista SA, Lu Z, Hofmann S, Freddo T, Overby DR, et al. (2008) Reduction of the available area for aqueous humor outflow and increase in meshwork herniations into collector channels following acute IOP elevation in bovine eyes. Invest Ophthalmol Vis Sci 49: 5346-5352.
- Lu Z, Overby DR, Scott PA, Freddo TF, Gong H (2008) The mechanism of increasing outflow facility by rho-kinase inhibition with Y-27632 in bovine eyes. Exp Eye Res 86: 271-281.
- Scott PA, et al. (2007) Comparative studies between species that do and do not exhibit the washout effect. Exp Eye Res 84: 435-443.
- Swaminathan SS, Oh DJ, Kang MH, Ren R, Jin R, et al. (2013) Secreted protein acidic and rich in cysteine (SPARC)-null mice exhibit more uniform outflow. Invest Ophthalmol Vis Sci 54: 2035-2047.
- Aga M, Bradley JM, Keller KE, Kelley MJ, Acott TS (2008) Specialized podosome- or invadopodia-like structures (PILS) for focal trabecular meshwork extracellular matrix turnover. Invest Ophthalmol Vis Sci 49: 5353-5365.
- Leske MC, Heijl A, Hussein M, Bengtsson B, Hyman L, et al. (2003) Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch Ophthalmol 121: 48-56.
- Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, et al. (2002) The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol 120: 714-720.
- Rohen JW, Lutjen-Drecoll E (1982) Biology of the trabecular meshwork. 1st ed. Basic Aspects of Glaucoma Research, ed. Lutjen-Drecoll E, Stuttgard: Schattauer FK, Verlap. 141-166.
- Roos H, Dahlberg L, Hoerrner LA, Lark MW, Thonar EJ, et al. (1995) Markers of cartilage matrix metabolism in human joint fluid and serum: the effect of exercise. Osteoarthritis Cartilage 3: 7-14.
- Rohen JW (1983) Why is intraocular pressure elevated in chronic simple glaucoma? Anatomical Considerations. Ophthalmology 90: 758-765.
- Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M, Grierson I (1993) Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG). Exp Eye Res 56: 683-692.
- Rohen JW, Schachtschabel DO, Wehrmann R (1982) Structural changes of human and monkey trabecular meshwork following in vitro cultivation. Graefes Arch Clin Exp Ophthalmol 218: 225-232.
- Lutjen-Drecoll E (2000) Importance of trabecular meshwork changes in the pathogenesis of primary open-angle glaucoma. J Glaucoma 9: 417-418.
- Lutjen-Drecoll E, Shimizu T, Rohrbach M, Rohen JW (1986) Quantitative analysis of “plaque material” in the inner- and outer wall of Schlemm’s canal in normal and glaucomatous eyes. Exp Eye Res 42: 443-455.
- Lutjen-Drecoll E, Tamm E, eds. (1987) Differences in the amount of ‘plaque-material’ in the outflow system of eyes with chronic simple and exfoliation glaucoma. Glaucoma Update III, ed. G.K. Krieglstein. Springer-Verlag: Berlin Heidelberg 17-22.
- Last JA, Pan T, Ding Y, Reilly CM, Keller K, et al. (2011) Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci 52: 2147-2152.
- Camras LJ, Stamer WD, Epstein D, Gonzalez P, Yuan F (2014) Circumferential tensile stiffness of glaucomatous trabecular meshwork. Invest Ophthalmol Vis Sci 55: 814-823.
- Alvarado J, Murphy C, Juster R (1984) Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology 91: 564-579.
- Alvarado J, Murphy C, Polansky J, Juster R (1981) Age-related changes in trabecular meshwork cellularity. Invest Ophthalmol Vis Sci 21: 714-727.
- Grierson I, Howes RC (1987) Age-related depletion of the cell population in the human trabecular meshwork. Eye (Lond) 1: 204-210.
- Dueker DK, Norberg M, Johnson DH, Tschumper RC, Feeney-Burns L (1990) Stimulation of cell division by argon and Nd-YAG laser trabeculoplasty in cynomolgus monkeys. Invest Ophthalmol Vis Sci 31: 115-124.
- Du Y, Roh DS, Mann MM, Funderburgh ML, Funderburgh JL, et al. (2012) Multipotent stem cells from trabecular meshwork become phagocytic TM cells. Invest Ophthalmol Vis Sci 53: 1566-1575.
- Kelley MJ, Rose AY, Keller KE, Hessle H, Samples JR, et al. (2009) Stem cells in the trabecular meshwork: present and future promises. Exp Eye Res 88: 747-751.
- Bhattacharya SK (2006) Focus on molecules: cochlin. Exp Eye Res 82: 355-356.
- Bhattacharya SK, Annangudi SP, Salomon RG, Kuchtey RW, Peachey NS, et al. (2005) Cochlin deposits in the trabecular meshwork of the glaucomatous DBA/2J mouse. Exp Eye Res 80: 741-744.
- Bhattacharya SK, Peachey NS, Crabb JW (2005) Cochlin and glaucoma: a mini-review. Vis Neurosci 22: 605-613.
- Bhattacharya SK, Rockwood EJ, Smith SD, Bonilha VL, Crabb JS, et al. (2005) Proteomics reveal Cochlin deposits associated with glaucomatous trabecular meshwork. J Biol Chem 280: 6080-6084.
- Gottanka J, Johnson DH, Martus P, Lutjen-Drecoll E (1997) Severity of optic nerve damage in eyes with POAG is correlated with changes in the trabecular meshwork. J Glaucoma 6: 123-132.
- Knepper PA, Goossens W, Hvizd M, Palmberg PF (1996) Glycosaminoglycans of the human trabecular meshwork in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 37: 1360-1367.
- Knepper PA, Goossens W, Palmberg PF (1996) Glycosaminoglycan stratification of the juxtacanalicular tissue in normal and primary open-angle glaucoma. Invest Ophthalmol Vis Sci 37: 2414-2425.
- Knepper PA, Goossens W, Mayanil CS (1998) CD44H localization in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 39: 673-680.
- Knepper PA, Mayanil CS, Goossens W, Wertz RD, Holgren C, et al. (2002) Aqueous humor in primary open-angle glaucoma contains increased levels of CD44S. Invest Ophthalmol Vis Sci 43: 133-139.
- Knepper PA, Goossens W, Mayanil CS (1998) CD44H localization in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 39: 673-680.
- Knepper PA, Mayanil CS, Goossens W, Wertz RD, Holgren C, et al. (2002) Aqueous humor in primary open-angle glaucoma contains an increased level of CD44S. Invest Ophthalmol Vis Sci 43: 133-139.
- Knepper PA, Miller AM, Choi J, Wertz RD, Nolan MJ, et al. (2005) Hypophosphorylation of aqueous humor sCD44 and primary open-angle glaucoma. Invest Ophthalmol Vis Sci 46: 2829-2837.
- Pattabiraman PP, Rao PV (2010) Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am J Physiol Cell Physiol 298: C749-C763.
- Rao PV, Deng PF, Kumar J, Epstein DL (2001) Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Invest Ophthalmol Vis Sci 42: 1029-1037.
- Rao PV, Deng P, Sasaki Y, Epstein DL (2005) Regulation of myosin light chain phosphorylation in the trabecular meshwork: role in aqueous humour outflow facility. Exp Eye Res 80: 197-206.
- Kaufman PL (2008) Enhancing trabecular outflow by disrupting the actin cytoskeleton, increasing uveoscleral outflow with prostaglandins, and understanding the pathophysiology of presbyopia interrogating Mother Nature: asking why, asking how, recognizing the signs, following the trail. Exp Eye Res 86: 3-17.
- Kaufman PL, Mittag TW (1994) Medical therapy of glaucoma., in Textbook of Ophthalmology, S.M. Podos, et al., Editors. Mosby: St Louis. p. 9.7-9.30.
- Clark AF, Brotchie D, Read AT, Hellberg P, English-Wright S, et al. (2005) Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton 60: 83-95.
- Clark AF, Miggans ST, Wilson K, Browder S, McCartney MD (1995) Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. J Glaucoma 4: 183-188.
- Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M, et al. (1994) Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci 35: 281-294.
- O’Reilly S, Pollock N, Currie L, Paraoan L, Clark AF, et al. (2011) Inducers of cross-linked actin networks in trabecular meshwork cells. Invest Ophthalmol Vis Sci 52: 7316-7324.
- Faralli JA, Schwinn MK, Gonzalez JM Jr, Filla MS, Peters DM (2009) Functional properties of fibronectin in the trabecular meshwork. Exp Eye Res 88: 689-693.
- Filla MS, Schwinn MK, Nosie AK, Clark RW, Peters DM (2011) Dexamethasone-associated cross-linked actin network formation in human trabecular meshwork cells involves β3 integrin signaling. Invest Ophthalmol Vis Sci 52: 2952-2959.
- Filla MS, Woods A, Kaufman PL, Peters DM (2006) Beta1 and beta3 integrins cooperate to induce syndecan-4-containing cross-linked actin networks in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 47: 1956-1967.
- Liu X, Hu Y, Filla MS, Gabelt BT, Peters DM, et al. (2005) The effect of C3 transgene expression on actin and cellular adhesions in cultured human trabecular meshwork cells and on outflow facility in organ cultured monkey eyes. Mol Vis 11: 1112-1121.
- Tan JC, Peters DM, Kaufman PL (2006) Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr Opin Ophthalmol 17: 168-174.
- Fuchshofer R, Tamm ER (2009) Modulation of extracellular matrix turnover in the trabecular meshwork. Exp Eye Res 88: 683-688.
- Fuchshofer R, Tamm ER (2012) The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res 347: 279-290.
- Fuchshofer R, Welge-Lussen U, Lutjen-Drecoll E (2003) The effect of TGF-beta2 on human trabecular meshwork extracellular proteolytic system. Exp Eye Res 77: 757-765.
- Fuchshofer R, Yu AH, Welge-Lussen U, Tamm ER (2007) Bone morphogenetic protein-7 is an antagonist of transforming growth factor-beta2 in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 48: 715-726.
- Braig S, Wallner S, Junglas B, Fuchshofer R, Bosserhoff AK (2011) CTGF is overexpressed in malignant melanoma and promotes cell invasion and migration. Br J Cancer 105: 231-238.
- Johnson D, Gottanka J, Flugel C, Hoffmann F, Futa R, et al. (1997) Ultrastructural changes in the trabecular meshwork of human eyes treated with corticosteroids. Arch Ophthalmol 115: 375-383.
- Clark AF, Wilson K, de Kater AW, Allingham RR, McCartney MD (1995) Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest Ophthalmol Vis Sci 36: 478-489.
- Fleenor DL, Shepard AR, Hellberg PE, Jacobson N, Pang IH, et al. (2006) TGFbeta2-induced changes in human trabecular meshwork: implications for intraocular pressure. Invest Ophthalmol Vis Sci 47: 226-234.
- Sethi A, Jain A, Zode GS, Wordinger RJ, Clark AF (2011) Role of TGFbeta/Smad signaling in gremlin induction of human trabecular meshwork extracellular matrix proteins. Invest Ophthalmol Vis Sci 52: 5251-5259.
- Wordinger RJ, Agarwal R, Talati M, Fuller J, Lambert W, et al. (2002) Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol Vis 8: 241-250.
- Wordinger RJ, Fleenor DL, Hellberg PE, Pang IH, Tovar TO, et al. (2007) Effects of TGF-beta2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Invest Ophthalmol Vis Sci 48: 1191-1200.
- Wordinger RJ, Sharma T, Clark AF (2014) The role of TGF-β2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J Ocul Pharmacol Ther 30: 154-162.
- Tektas OY, Lutjen-Drecoll E (2009) Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res 88: 769-775.
- Borras T (2003) Gene expression in the trabecular meshwork and the influence of intraocular pressure. Prog Retin Eye Res 22: 435-463.
- Borras T (2003) Recent developments in ocular gene therapy. Exp Eye Res 76: 643-652.
- Borras T, Brandt CR, Nickells R, Ritch R, et al. (2002) Gene therapy for glaucoma: treating a multifaceted, chronic disease. Invest Ophthalmol Vis Sci 43: 2513-2518.
- Xue W, Wallin R, Olmsted-Davis EA, Borras T (2006) Matrix GLA protein function in human trabecular meshwork cells: inhibition of BMP2-induced calcification process. Invest Ophthalmol Vis Sci 47: 997-1007.