
Research Article
*Address for Correspondence: Yolanda Lorenzo PhD, Center for Eye Research, Department of Ophthalmology, Oslo University Hospital and University of Oslo, Pb 4956 Nydalen, 0424 Oslo, Norway, Tel: +47-45459663; Fax: +47-22119989; E-mail: y.c.lorenzo@medisin.uio.no
Citation: Lorenzo Y, Berg KH, Ustgaard-Andersen K, Johnsen EO, Ringvold A. Trypsin for Dissociation of Limbal Cells for Engineering of Grafts May Induce DNA Strand Breaks in the Harvested Cells. J Ocular Biol. 2013;1(2): 6.
Copyright © 2013 Lorenzo Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 1, Issue: 2
Submission: 28 September 2013 | Accepted: 06 November 2013 | Published: 11 November 2013

 Incubation of samples subjected to enzyme dissociation in culture medium revealed that incubation using 0.05% trypsin-EDTA for 1 and also for 3 hours failed to release part of the epithelial cell population. After 14 to 16 days confluent zones were produced in all of the 32 cultures. Although variation in extent of outgrowth was observed, all the samples produced confluent cultures extending more than 5 mm from the limbal cut edge at each side of the sample (Figure 4).
Incubation of samples subjected to enzyme dissociation in culture medium revealed that incubation using 0.05% trypsin-EDTA for 1 and also for 3 hours failed to release part of the epithelial cell population. After 14 to 16 days confluent zones were produced in all of the 32 cultures. Although variation in extent of outgrowth was observed, all the samples produced confluent cultures extending more than 5 mm from the limbal cut edge at each side of the sample (Figure 4).

Trypsin for Dissociation of Limbal Cells for Engineering of Grafts May Induce DNA Strand Breaks in the Harvested Cells
Yolanda Lorenzo1*, Kristiane Haug Berg1, Kristine Ustgaard-Andersen1, Erik Otter Johnsen1, Amund Ringvold1,2, Morten C. Moe1,2, Liv Drolsum1,2, Katerina Jirsova4, Bjørn Nicolaissen1,2 and Andrew Collins3
- 1Center for Eye Research, Department of Ophthalmology, Oslo University Hospital, Oslo, Norway
- 2Institute for Clinical Medicine, University of Oslo, Oslo, Norway
- 3Department of Nutrition, Institute for Basic Medical Sciences, University of Oslo, Oslo, Norway
- 4Laboratory of the Biology and Pathology of the Eye, Institute of Inherited Metabolic Disorders, Charles University in Prague, Prague, Czech Republic
*Address for Correspondence: Yolanda Lorenzo PhD, Center for Eye Research, Department of Ophthalmology, Oslo University Hospital and University of Oslo, Pb 4956 Nydalen, 0424 Oslo, Norway, Tel: +47-45459663; Fax: +47-22119989; E-mail: y.c.lorenzo@medisin.uio.no
Citation: Lorenzo Y, Berg KH, Ustgaard-Andersen K, Johnsen EO, Ringvold A. Trypsin for Dissociation of Limbal Cells for Engineering of Grafts May Induce DNA Strand Breaks in the Harvested Cells. J Ocular Biol. 2013;1(2): 6.
Copyright © 2013 Lorenzo Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 1, Issue: 2
Submission: 28 September 2013 | Accepted: 06 November 2013 | Published: 11 November 2013
Abstract
Aim: Cultures for engineering of transplantable limbal epithelial grafts for treatment of ocular surface disorders may be initiated using dissociation of limbal epithelial cells by trypsin-EDTA or dispase or by a sequential incubation with these enzymes. The safety of such procedures is debated, and in the present study we examined levels of DNA damage in cells dissociated by a commonly used concentration of trypsin. Limbal samples subjected to the dissociation procedure were subsequently cultivated and monitored for outgrowth of cells.Methods: Corneo-limbal rings were retrieved after transplant surgery, divided into samples measuring approx. 2x2 mm (n=32), and incubated in 0.05% trypsin-EDTA for one or three hours in either 250 μl or in 3 ml of the enzyme solution at 37 °C. DNA damage (strand breaks plus alkali-labile sites) was assessed using single cell gel electrophoresis (Comet assay) and evaluation of tail intensity (TI). Outgrowths from the cultivated samples were monitored by phase contrast microscopy and cells were subjected to Hoechst.
Results: Noticeable levels of DNA damage were seen regardless of incubation time and volume of enzyme solution. There was a trend towards increased levels of damage in cells when using 3 ml compared to values recorded in cells dissociated in 250 μl of the enzyme solution. Outgrowth of cells was observed from all of the 32 cultivated samples.
Conclusion: Dissociation of human limbal epithelial cells by a commonly used concentration of trypsin-EDTA may induce evident DNA damage in the cell population destined for graft production. The current methods for cell dissociation should be examined more closely for induction of damage to essential molecular constituents of the cells including to the stem cell population. Procedural steps and components of the ex vivo system that may reduce such damage and/or facilitate repair should be identified.
Introduction
Corneal transparency and vision depend on intactness and proper functioning of the corneal epithelium and the limbal epithelial stem cell population. The stem cells are localized in the basal layer of the limbal epithelium and also in the limbal crypts, and continuous renewal of the corneal epithelium relies critically on the health of the stem cell population [1,2]. A number of conditions and ocular insults may adversely affect the regenerative potential of these limbal stem cells and induce a condition called limbal stem cell deficiency (LSCD). Initially, LSCD is characterized by recurrent epithelial defects and ocular pain. Ultimately, the normal transparent corneal epithelium is replaced by vascularized and inflamed conjunctival tissue and the loss of vision is severe [3]. For these patients, the traditional corneal transplant procedure is not an option. The corneal surface represents a hostile microenvironment, and a penetrating donor graft is regularly subjected to immune rejection despite immune-modulatory therapy.For patients with LSCD, developments within the fields of tissue engineering have provided new treatment options. Using samples of healthy limbal tissue, Pellegrini et al. and Schwab produced epithelial grafts ex vivo and transplanted such tissues to diseased eyes after removal of the pathological corneal surface [4,5].
The procedure was found to improve or to restore vision in a majority of the cases, and subsequent studies from other groups confirmed the potential of this new therapeutic approach [6-12].
In the first protocol designed by Pellegrini et al., the limbal samples were incubated in 0.05% trypsin-EDTA at 37 °C for 3 hours, and the dissociated cells were seeded on a feeder layer of 3T3 cells in a medium containing fetal bovine serum (FBS), a mixture of growth factors and hormones, and also cholera toxin [4]. Reportedly, such a protocol regularly secures that more than 3% of the cells in the generated grafts are positive for p63. Content of p63+ cells above this level is associated with improved long term outcome after transplantation [13].
Despite such information, current laboratory procedures for engineering of grafts differ between clinics. For initiation of cultures, the epithelial cells may be dissociated by trypsin-EDTA [13-15], by dispase [16,17], or by a sequential combination of dispase and trypsin-EDTA with or without accompanying mechanical procedures [12]. In order to expand the dissociated cells ex vivo, however, most protocols include use of a medium supplemented with a mixture of hormones, growth factors, cholera toxin and also FBS.
An alternative approach is the explant culture technique [11]. Here the limbal samples are positioned in medium on a suitable substrate without initial dissociation of the epithelial cells. In recent studies, using this approach, we have demonstrated that transplantable grafts may be generated in a culture system without animal feeder cells and FBS using autologous serum as the single growth promoting supplement [18,19].
A number of experimental studies have examined the effect of dissociation procedures on various critical parameters such as cell membrane integrity, cell yield, colony forming efficiency (CEF), proliferative potential, and percentage of cells presenting markers associated with stemness [20-27]. While optimal results for clinical purposes reportedly depend on adherence to the above outlined protocol [4], the results from experimental studies may indicate an adverse effect of trypsin-EDTA on viability [25], and on the proliferative potential of the dissociated cells [23,27], and such dissociation may also render the cells dependent on animal feeder cells for proper colony forming efficiency.
From a clinical point of view, the content of non-human additives and animal cells in a culture system designed for ex vivo generation of tissues for transplantation to human patients may increase the risk of immune rejection and transfer of animal pathogens [19]. The above mentioned experimental studies may indicate, that such a complex culture system is needed in order to support repair of damage inflicted upon essential cellular molecules during the dissociation procedure.
Maintained integrity of cellular and DNA repair mechanisms is essential for proper cellular functioning including for long term viability and proliferative potential. In animal eyes a gradient of DNA damage has been described in the corneal epithelium. Levels of DNA strand breaks was observed to decrease significantly from the surface layers towards the basal layer [28]. Scant information is available about DNA damage in human corneal and limbal epithelium. In a recent study we found the levels of DNA strand breaks to be very low in corneo-limbal epithelial cells after storage in Optisol GS close to the upper recommended limit [29]. Transfer of such samples to culture was associated with proliferative activity and expression of markers characterizing differentiated as well as undifferentiated cells in the limbal epithelium, but also with an increase in the levels of DNA strand breaks.
The effect of enzymatic dissociation on DNA in the harvested limbal epithelial cells has not previously been examined. In this first study our aim is to examine limbal epithelial cells for DNA damage subsequent to dissociation of cells by incubation in 0.05% trypsin-EDTA at 37 °C for 1 hour, an interval previously used for dissociation of cells from organ cultured tissues [27], and for 3 hours routinely used for epithelial dissociation of cells from fresh samples [4]. Information on volume of the enzyme solution is generally not provided in the published protocols. For incubation of samples measuring approximately 2x2 mm, based on pilot experiments, the samples were incubated in either 250 μl or 3 ml of the solution. Routinely, the tissues subjected to incubation were cultivated in order to examine for outgrowth of cells.
 All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
 Cell dissociation
Cell dissociation
Material and Methods
All experiments were conducted in accordance with the Declaration of Helsinki and all tissue harvesting was approved by the Local Committees for Medical Research Ethics.Tissue
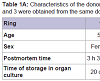
Human corneo-scleral tissue was obtained from rings available after penetrating keratoplasty and preserved in organ culture prior to use. For age, sex, postmortem time, and time in organ culture, see Table 1A.
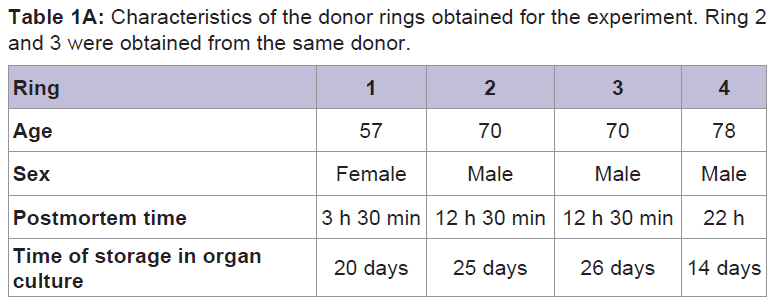
Table 1A: Characteristics of the donor rings obtained for the experiment. Ring 2 and 3 were obtained from the same donor.
The corneo-limbal rings were transferred to a 10 mm dishes (Nunclon Surface, Nunc, Denmark) with 15 ml of DMEM/F12 (Invitrogen). Peripheral sclera and cornea were trimmed off, and the rings were divided into 12 samples that measured approximately 2x2 mm. Samples were washed 3x5 min in 15 ml Hanks Balanced Salt Solution without Ca2+ and Mg2+ (HBSS) at room temperature (Figure 1A).
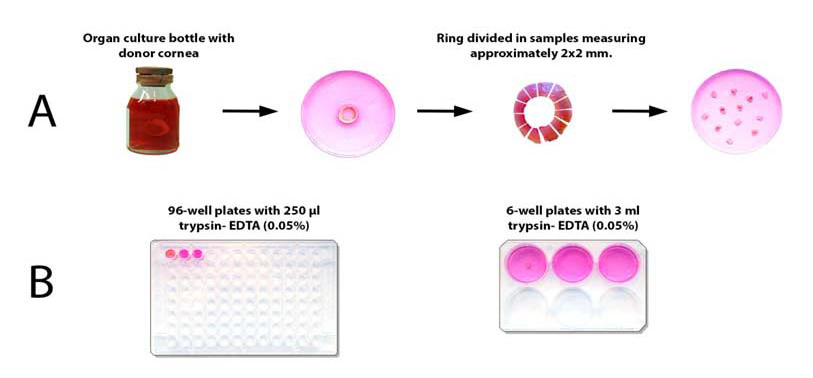
Figure 1: Four human corneal- limbal rings were obtained after corneal transplant surgery. The rings were divided in 12 samples (A). Duplicate samples were incubated individually at 37 °C for 1 or 3 hours in either 250 μl or 3 ml trypsin-EDTA (0.05%) using 96-well plates or 6-well plates respectively (B).
Duplicate samples from each ring were incubated at 37 °C in a humid atmosphere containing 5% CO2 in pre-equilibrated 0.05% trypsin in HBSS with 0.02% EDTA-4Na and without Ca2+ and Mg2+ for one or three hours (n=32) in 250 μl or in 3 ml of the solution using 96-well plates (Nunclon Surface, Nunc, Denmark) or 6-well plates (PerkinElmer Life Sciences) respectively (Figure 1B). At the end of incubation, enzyme activity was terminated by adding an equal amount of a growth medium DMEM/F12 containing serum (see below). Cells were dispersed by gentle pipetting x 20 using a 1000 μl micropipette for samples incubated in 96-well plates and a 3 ml plastic pipette for samples incubated in 6-well plates. The dissociated cells in medium/enzyme solution from each well were transferred to tubes on ice and processed for DNA damage analysis.
Comet assay for evaluation of DNA damage
Using a standard comet assay protocol, the basal levels of strand breaks (SBs) were measured as described [30]. For procedural control, human lymphocytes were used. Cells were resuspended in PBS at 0.25x106 cells/ml. 20 μl of cell suspension was mixed with 94 μl of 1% low-melting point agarose, and 5 μl drops were placed onto agarose-precoated slides following a format of 12 minigels/slide [31]. Cells were lysed in 2.5 M NaCl, 0.1 M EDTA, 0.01 M Tris and 1% Triton X-100 (pH 10) at 4 °C for at least 1 h. After that, slides were immersed in electrophoresis solution (0.3 M NaOH and 1 mM EDTA) for 20 min. Electrophoresis was then carried out at 1.3 V/cm for 20 min in the same solution. Slides were washed for 10 min in PBS, 10 min in water, fixed in 70% ethanol for 15 min and in absolute ethanol for a further 15 min. Comets were stained with SYBR Gold at the dilution recommended by the manufacturer in a large dish at 4 °C with agitation. Using a fluorescence microscope we visually classified 100 comets (50 on each gel) into 5 categories, 0-4, representing increasing relative tail intensities. Summing the scores (0-4) of 100 comets therefore gives an overall score of between 0 and 400 arbitrary units [31].
Cultivation and examination of outgrowth from limbal tissue
The limbal samples subjected to the various protocols for cell dissociation were each positioned epithelial side down in tissue culture coated 6 well dishes containing 1.5 ml medium DMEM/F12 (Invitrogen) supplemented with 5% FBS, epidermal growth factor (EGF)(2 ng/ml, R&D Systems, MN), insulin 5 μg/ml, transferrin 5 μg/ml and sodium selenite 5 ng/ml (ITS), cholera toxin A (30 ng/ml, Biomol International, LP), dimethylsulphoxide (DMSO,0.5%), hydrocortisone (15 μM), gentamicin (50 μg/ml), Penicillin/Streptomycin (100 U/ml), amphotericin B (2.5 μg/ml), and incubated at 37 °C in a humid atmosphere containing 5% CO2. After one to two days 1.5 ml medium was added, and medium was then changed every second or third day up to 14-16 days.
Cultures were monitored by phase contrast microscopy; micrographs were obtained using a Nikon eclipse TS100 microscope.
After 14 to 16 days, the cultured cells were fixed in 4% formaldehyde, washed in PBS, and stored at 4 °C for evaluation.
Cultures were either left unstained or subjected to immunohistochemistry for detection of cells positive for p63 Ab-4 (Clone 4A4+Y4A3, 1:1600, Thermo Scientific). The positive immunoreaction of the primary antibody was detected by a secondary antibody conjugated with the fluorescent marker Alexa Fluor 488 donkey anti mouse Ig (Cat # A 21202, 1:500, Invitrogen). Hoechst (1:500, Invitrogen) was used for nuclear staining.
Results
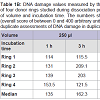
DNA damage, in form of SBs was observed after cell dissociation regardless of incubation time and volume. The levels of DNA damage were lower in all samples when incubation was performed using 250 μl of enzyme solution in 96-well plates compared with levels recorded when using 3 ml in 6-well plates. In some cases increasing the incubation time when using the same volume did not cause any overt increase in the levels of DNA damage (Table 1B, Figure 2, Figure 3).The DNA median value, in form of SBs, in the lymphocytes used as a control of the procedure was 10 arbitrary units.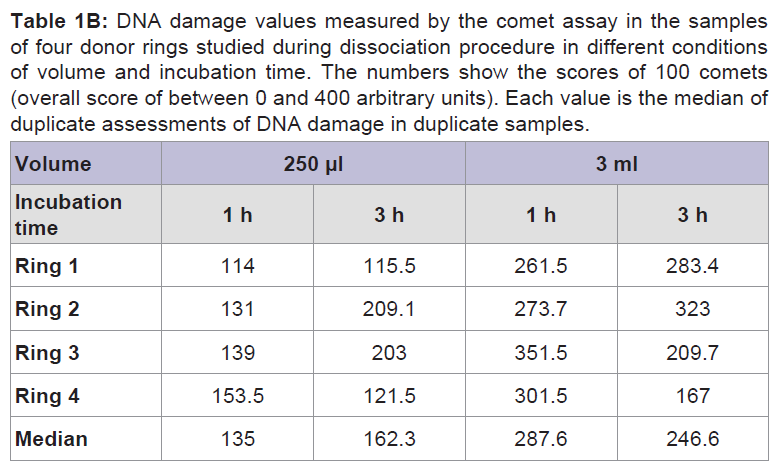
Table 1B: DNA damage values measured by the comet assay in the samples of four donor rings studied during dissociation procedure in different conditions of volume and incubation time. The numbers show the scores of 100 comets (overall score of between 0 and 400 arbitrary units). Each value is the median of duplicate assessments of DNA damage in duplicate samples.
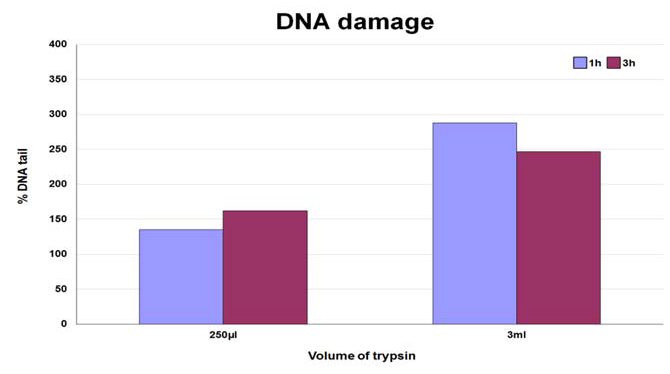
Figure 2: DNA damage in the form of SBs in cells dissociated with trypsin in 250 μl in 96-well plates or in 3 ml in 6-well plates for 1 or 3 hours.
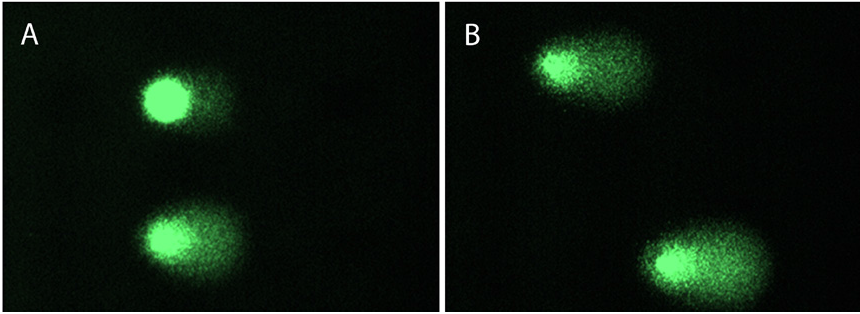
Figure 3: DNA damage visualised with comet assay after dissociation procedure of samples with trypsin (A) in 96-well plates or (B) in 6-well plates.
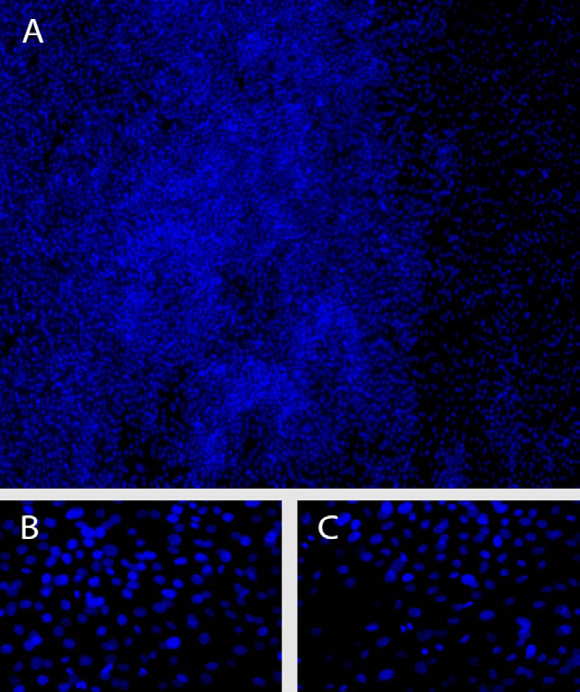
Figure 4: A) Photo of outgrowth from the limbal cut edge in samples subjected to enzyme dissociation. Areas with high density of nuclei were regularly observed (50X). B) Photo of outgrowth close to the limbal edge (400X). C) Photo 5 mm outside of the limbal edge (400X).
Discussion
The present study demonstrates for the first time, that dissociation of cells from the corneo-limbal epithelium using 0.05% trypsin-EDTA at 37 °C is associated with noticeable levels of DNA damage in the harvested cells. DNA damage was observed regardless of incubation time and volume of enzyme solution. There was a trend towards higher levels of strand breaks when incubating the samples in 3 ml compared to values recorded in cells dissociated in 250 μl.In patients with one healthy eye, fresh autologous limbal tissue may be harvested for ex vivo generation of grafts. In cases with bilateral LSCD, limbal donor tissue may be obtained from an Eye Bank. For experimental studies on the human limbal epithelium, the tissues are - with few exceptions - derived from donor corneas that have been subjected to Eye Bank storage using a cold preservation or an organ culture system. In both systems, cell death is observed during prolonged storage and a number of parameters may influence the viability and proliferative potential of the epithelial tissue and also increase the sensitivity of the cells towards insults [32-35].
The results obtained in the present study may therefore not apply to freshly harvested and dissociated cells. Although the culture conditions may contribute to some levels of DNA damage [29], the findings in previous experimental studies, and in the present, underline the need for a continuous critical examination of the effect of the various dissociation protocols on essential molecular constituents of the harvested cells.
Viability of the epithelial tissue in samples subjected to dissociation is documented by the outgrowth of cells from all samples after transfer to culture dishes and incubated in SHEM medium at 37 °C and 5% CO2. This is in line with findings reported by Xie et al. [22,26]. In their studies, dissociation using dispase at 4 °C failed to detach a substantial number of limbal cells including cells positive for markers associated with stemness.
Using organ cultured donor corneo-limbal tissue, Zito-Abbad et al. examined the effect of incubation in 1.2 IU/ml dispase II and in a 0.05% trypsin-0.01% EDTA solution at 37 °C for 1 hour. Age of the donors, time from death to corneal retrieval, and also time in organ culture is similar to those in the present study [27]. They found that incubation of samples in trypsin-EDTA, but not in dispase, significantly decreased cell proliferation at two and three weeks. Further, the duration of corneal organ culture did not influence the cell growth. Kim et al. dissociated limbal cells from corneo-sceral rings obtained after five days of cold storage using 0.05% trypsin-0.01% EDTA at 37 °C with gentle agitation and observed that a CFE of 6.63±2.35% was maintained until passage four. The discrepant results may be due to source of tissue and storage procedure prior to dissociation, and also to differences in composition of culture medium and type of feeder cells. The latter group used DMEM/F12 supplemented with 10% FBS, 10 ng/ml EGF, 5 μg/ml insulin, 0.1 nM choleratoxin, 50 IU/ml penicillin-streptomycin, 0.18 mM adenine, 4 mM glutamine, 0.4 μg/ml hydrocortisone, and 2 nM triiodothyronine (SHEM) and 3T3 feeder cells. By Lindberg et al. [36], this culture system was shown to support a mean of 23 population doublings prior to senescence after dissociation of the cells in a 1:1 mixture of 0.1% trypsin and 0.02% EDTA in isotonic phosphate-buffered saline. Slightly modified, these culture conditions were adapted by Pellegrini et al. [4].
Similarly, divergent results have been obtained in studies using increased concentration of trypsin for dissociation. In a comprehensive study Meyer-Blazejewska et al. examining the effect of various dissociation methods on biopsies obtained from donor corneas. For trypsin-EDTA, the concentrations were 0.25% trypsin-0.02% EDTA and the samples were minced and incubated at 37°C for 1.5 hours under continuous agitation or magnetic stirring. Specimens were also incubated in a 2.4 U/ml dispase II solution at 37 °C for 1.5 hours followed by incubation in 0.25% trypsin-0.02% EDTA at 37 °C for 10 minutes. In this study, the combined dispase II/trypsin-EDTA dissociation procedure yielded a consistently high number of viable cells (85%-90%) than the single step incubation in 0.25% trypsin-EDTA with agitation. When seeded on 3T3 feeder cells, the CFE for dispase II/trypsin-EDTA was 0.37%, for trypsin-EDTA with magnetic stirring 0.35%, and for trypsin-EDTA with agitation 0.34%. Colony density and size were highest with a combined extraction method using dispase II/trypsin-EDTA and a single extraction method using trypsin-EDTA with agitation. However, the culture media tested differed in composition from SHEM.
In contrast, Arpitha et al. showed that a sequential incubation of limbal tissues in 0.25% trypsin in Ca2/Mg2 free phosphate buffered saline at 37 °C for 50 min and subsequently in 2 mg/ml dispase II in DMEM at 37 °C for 30 min yielded a cell population with a significant increase in density of cells positive for p63, a high percentage of intact cells when tested with the trypan blue dye exclusion test (mean viability of 93.2% ± 6.2%), and also a high capacity for generation of large colonies with a compact morphology when cultivated in SHEM medium on 3T3 feeder cells [20].
Finally, Chen SY et al. provided evidence, that dissociation of cells from sheets or clusters by incubation in 0.25% trypsin and 1 mM EDTA at 37 °C for 15 min may render the isolated cells dependent on 3T3 feeder layers for maintained clonal formation capacity [21].
Results from experimental studies, including the present, may indicate that varying results in assays using trypsin-EDTA for dissociation may in part be related to the ability of the ex vivo system including composition of medium and presence of feeder cells to support repair of molecular damage inflicted upon the cells by the dissociation procedure.
Tissue engineering provides a potent novel tool for treatment of ocular disorders and the clinical results are promising. However, the procedures are still new and in their shaping. Results from experimental studies, including the present, emphasize the need for evaluation of the various steps included in commonly used protocols in order to improve the procedures and safeguard the quality of the tissue generated for transplantation.
Acknowledgements
The study was supported by grants from Blindemissionen IL, The Norwegian Association of the Blind and Partially Sighted, The Research Council of Norway, The Faculty of Medicine University of Oslo and Oslo University Hospital. KJ was supported by PRVOUK-P24/LF1/3 programs of the Charles University in Prague.The authors would like to thank Geir Qvale, Linn Lillevold and Eli Gulliksen (Center for Eye Research, OUS) and Kahsai Beraki (Dept. of Pathology, OUS) for their excellent technical assistance and support.References
- Cotsarelis G, Cheng SZ, Dong G, Sun TT, Lavker RM (1989) Existence of slow-cycling limbal epithelial basal cells that can be preferentially stimulated to proliferate: implications on epithelial stem cells. Cell 57: 201-209.
- Davanger M, Evensen A (1971) Role of the pericorneal papillary structure in renewal of corneal epithelium. Nature 229: 560-561.
- Tseng SC (1996) Regulation and clinical implications of corneal epithelial stem cells. Mol Biol Rep 23: 47-58.
- Pellegrini G, Traverso CE, Franzi AT, Zingirian M, Cancedda R, et al. (1997) Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet 349: 990-993.
- Schwab IR (1999) Cultured corneal epithelia for ocular surface disease. Trans Am Ophthalmol Soc 97: 891-986.
- Daya SM, Ilari FA (2001) Living related conjunctival limbal allograft for the treatment of stem cell deficiency. Ophthalmology 108: 126-133.
- Di Girolamo N, Bosch M, Zamora K, Coroneo MT, Wakefield D, et al. (2009) A contact lens-based technique for expansion and transplantation of autologous epithelial progenitors for ocular surface reconstruction. Transplantation 87: 1571-1578.
- Kolli S, Ahmad S, Lako M, Figueiredo F (2010) Successful clinical implementation of corneal epithelial stem cell therapy for treatment of unilateral limbal stem cell deficiency. Stem Cells 28: 597-610.
- Mariappan I, Maddileti S, Savy S, Tiwari S, Gaddipati S, et al. (2010) In vitro culture and expansion of human limbal epithelial cells. Nat Protoc 5: 1470-1479.
- Meller D, Pauklin M, Westekemper H, Steuhl KP (2010) Autologous transplantation of cultivated limbal epithelium. Ophthalmologe 107: 1133-1138.
- Sangwan VS, Basu S, Vemuganti GK, Sejpal K, Subramaniam SV, et al. (2011) Clinical outcomes of xeno-free autologous cultivated limbal epithelial transplantation: a 10-year study. Br J Ophthalmol 95: 1525-1529.
- Shortt AJ, Secker GA, Rajan MS, Meligonis G, Dart JK, et al. (2008) ex vivo expansion and transplantation of limbal epithelial stem cells. Ophthalmology 115: 1989-1997.
- Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, et al. (2010) Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med 363: 147-155.
- Harkin DG, Barnard Z, Gillies P, Ainscough SL, Apel AJ (2004) Analysis of p63 and cytokeratin expression in a cultivated limbal autograft used in the treatment of limbal stem cell deficiency. Br J Ophthalmol 88: 1154-1158.
- Marchini G, Pedrotti E, Pedrotti M, Barbaro V, Di Iotio E, et al. (2012) Long-term effectiveness of autologous cultured limbal stem cell grafts in patients with limbal stem cell deficiency due to chemical burns. Clin Experiment Ophthalmol 40: 255-267.
- Nakamura T, Inatomi T, Sotozono C, Ang LP, Koizumi N, et al. (2006) Transplantation of autologous serum-derived cultivated corneal epithelial equivalents for the treatment of severe ocular surface disease. Ophthalmology 113: 1765-1772.
- Prabhasawat P, Ekpo P, Uiprasertkul M, Chotikavanich S, Tesavibul N (2012) Efficacy of cultivated corneal epithelial stem cells for ocular surface reconstruction. Clin Ophthalmol 6: 1483-1492.
- Pathak M, Cholidis S, Haug K, Shahdadfar A, Moe MC, et al. (2012) Clinical transplantation of ex vivo expanded autologous limbal epithelial cells using a culture medium with human serum as single supplement: a retrospective case series. Acta Ophthalmol.
- Shahdadfar A, Haug K, Pathak M, Drolsum L, Olstad OK, et al. (2012) Ex vivo expanded autologous limbal epithelial cells on amniotic membrane using a culture medium with human serum as single supplement. Exp Eye Res 97: 1-9.
- Arpitha P, Prajna NV, Srinivasan M, Muthukkaruppan V (2008) A method to isolate human limbal basal cells enriched for a subset of epithelial cells with a large nucleus/cytoplasm ratio expressing high levels of p63. Microsc Res Tech 71: 469-476.
- Chen SY, Hayashida Y, Chen MY, Xie HT, Tseng SC (2011) A new isolation method of human limbal progenitor cells by maintaining close association with their niche cells. Tissue Eng Part C Methods 17: 537-548.
- Xie HT, Chen SY, Li GG, Tseng SC (2011) Limbal epithelial stem/progenitor cells attract stromal niche cells by SDF-1/CXCR4 signaling to prevent differentiation. Stem Cells 29: 1874-1885.
- Kawakita T, Shimmura S, Higa K, Espana EM, He H, et al. (2009) Greater growth potential of p63-positive epithelial cell clusters maintained in human limbal epithelial sheets. Invest Ophthalmol Vis Sci 50: 4611-4617.
- Kim MK, Lee JL, Shin KS, Jung GA, Wee WR, et al. (2006) Isolation of putative corneal epithelial stem cells from cultured limbal tissue. Korean J Ophthalmol 20: 55-61.
- Meyer-Blazejewska EA, Kruse FE, Bitterer K, Meyer C, Hofmann-Rummelt C, et al. (2010) Preservation of the limbal stem cell phenotype by appropriate culture techniques. Invest Ophthalmol Vis Sci 51: 765-774.
- Xie HT, Chen SY, Li GG, Tseng SC (2012) Isolation and expansion of human limbal stromal niche cells. Invest Ophthalmol Vis Sci 53: 279-286.
- Zito-Abbad E, Borderie VM, Baudrimont M, Bourcier T, Laroche L, et al. (2006) Corneal epithelial cultures generated from organ-cultured limbal tissue: factors influencing epithelial cell growth. Curr Eye Res 31: 391-399.
- Choy CK, Benzie IF, Cho P (2005) UV-mediated DNA strand breaks in corneal epithelial cells assessed using the comet assay procedure. Photochem Photobiol 81: 493-497.
- Haug K, Azqueta A, Johnsen-Soriano S, Shahdadfar A, Drolsum LK, et al. (2013) Donor cornea transfer from Optisol GS to organ culture storage: a two-step procedure to increase donor tissue lifespan. Acta Ophthalmol 91: 219-225.
- Collins AR (2004) The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 26: 249-261.
- Shaposhnikov S, Azqueta A, Henriksson S, Meier S, Gaivao I, et al. (2010) Twelve-gel slide format optimised for comet assay and fluorescent in situ hybridisation. Toxicol Lett 195: 31-34.
- Crewe JM, Armitage WJ (2001) Integrity of epithelium and endothelium in organ-cultured human corneas. Invest Ophthalmol Vis Sci 42: 1757-1761.
- Komuro A, Hodge DO, Gores GJ, Bourne WM (1999) Cell death during corneal storage at 4 degrees C. Invest Ophthalmol Vis Sci 40: 2827-2832.
- Joseph A, Powell-Richards AO, Shanmuganathan VA, Dua HS (2004) Epithelial cell characteristics of cultured human limbal explants. Br J Ophthalmol 88: 393-398.
- Shanmuganathan VA, Rotchford AP, Tullo AB, Joseph A, Zambrano I, et al. (2006) Epithelial proliferative potential of organ cultured corneoscleral rims; implications for allo-limbal transplantation and eye banking. Br J Ophthalmol 90: 55-58.
- Lindberg K, Brown ME, Chaves HV, Kenyon KR, Rheinwald JG (1993) In vitro propagation of human ocular surface epithelial cells for transplantation. Invest Ophthalmol Vis Sci 34: 2672-2679.