
Journal of Oncobiomarkers
Download PDF
Review Article
Address for Correspondence: Ming-Zhi Zhang, S-3206, MCN, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA, Phone: 615-343-1548; Fax: 615-343-2675; E-mail: ming-zhi.zhang@vanderbilt.edu
Citation: Yang S, Jiang L, Zhang MZ. 11β-Hydroxysteroid Dehydrogenase Type II is a Potential Target for Prevention of Colorectal Tumorigenesis. J Oncobiomarkers. 2013;1(1): 8.
Copyright © 2013 Shilin Yang, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oncobiomarkers | ISSN: 2325-2340 | Volume: 1, Issue: 1

11β-Hydroxysteroid Dehydrogenase Type II is a Potential Target for Prevention of Colorectal Tumorigenesis
Shilin Yang 1 , Li Jiang 1 and Ming-Zhi Zhang 1,2
- 1Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
- 2Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN, USA
Address for Correspondence: Ming-Zhi Zhang, S-3206, MCN, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA, Phone: 615-343-1548; Fax: 615-343-2675; E-mail: ming-zhi.zhang@vanderbilt.edu
Citation: Yang S, Jiang L, Zhang MZ. 11β-Hydroxysteroid Dehydrogenase Type II is a Potential Target for Prevention of Colorectal Tumorigenesis. J Oncobiomarkers. 2013;1(1): 8.
Copyright © 2013 Shilin Yang, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oncobiomarkers | ISSN: 2325-2340 | Volume: 1, Issue: 1
Abstract
Colorectal cancer (CRC) is a leading cause of cancer death, yet primary prevention remains the best approach to reducing overall morbidity and mortality. There is a clear molecular link between cyclooxygenase-2 (COX-2)-derived prostaglandin E2 (PGE2) production and CRC progression. Although selective COX-2 inhibitors as well as non-steroidal anti-inflammatory drugs (NSAIDs) reduce the number and sizes of colonic adenomas, increased cardiovascular risks of selective COX-2 inhibitors and increased gastrointestinal side-effects of NSAIDs limit their use in chemoprevention of CRC. Glucocorticoids induce apoptosis and are endogenous, potent COX-2 inhibitors. Glucocorticoids have been used for the treatment of hematologic malignancies, but not for solid tumors due to adverse side-effects such as immunosuppression and osteoporosis. In tissues, glucocorticoid actions are down-regulated by type 2 11β-hydroxysteroid dehydrogenase (11βHSD2), and inhibition of 11βHSD2 activity will elevate intracellular active glucocorticoid to levels that effectively suppress COX-2 expression. Both COX-2 and 11βHSD2 increase in Apc+/min mouse intestinal adenomas and human colonic adenomas and either pharmacologic or genetic 11βHSD2 inhibition leads to decreases in COX-2-mediated PGE2 production in tumors and prevents adenoma formation, tumor growth, and metastasis. 11βHSD2 inhibition may represent a novel approach for CRC chemoprevention by increasing tumor cell intracellular glucocorticoid activity, which in turn inhibits tumor growth by suppressing the COX-2-derived PGE2 pathway, as well as other pathways, without potential side-effects relating to chronic application of COX-2 inhibitors, NSAIDs and glucocorticoids.Introduction
Colorectal cancer (CRC) is ranked the third most common form of cancer worldwide in terms of incidence and mortality [1]. Most patients present with advanced disease, and metastatic disease remains largely incurable. Therefore, prevention remains the best approach to reduce the overall morbidity and mortality of CRC.In 1950, the Nobel Prize for Physiology or Medicine was awarded to Tadeus Reichstein, Edward C. Kendall and Philip S. Hench for the isolation of glucocorticoid hormones and the discovery that cortisol dramatically alleviated symptoms of rheumatoid arthritis [2]. Glucocorticoids are produced in the zona fasciculata (middle cortical layer) of adrenal gland. Glucocorticoids regulate a variety of important cardiovascular, metabolic, immunologic, and homeostatic functions and are important for fetal development. Glucocorticoids have been used to treat diseases caused by an overactive immune system, such as allergies, asthma, and autoimmune diseases as well as a broad spectrum of hematologic malignancies, including leukemia, lymphoma, and myeloma due to their ability to induce apoptosis [3-5]. Application of glucocorticoids in solid tumors is limited due to adverse side effects such as immunosuppression, inhibition of bone formation, suppression of calcium absorption and delayed wound healing.
Regulation of Glucocorticoid Actions
Although circulating levels of glucocorticoids are much higher than most hormones, only about 10% of circulating glucocorticoid is free, because most of circulating glucocorticoids bind to plasma proteins, particularly the corticosteroid-binding globulin, transcortin. As protein-bound glucocorticoid is biologically inactive, protein binding leads to decreases in metabolic clear rate and attenuation of fluctuations of glucocorticoid concentration.Circulating glucocorticoid levels are regulated by the hypothalamo-pituitary-adrenal axis [6]. Peptide corticotropinreleasing hormone (CRH) released from hypothalamus following stressors stimulates the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which in turn stimulates adrenal gland to secrete glucocorticoids. When blood concentrations of glucocorticoids rise above a certain threshold, they inhibit CRH secretion from the hypothalamus, leading to turning off of ACTH secretion and subsequent turning off of glucocorticoid secretion from adrenal gland.
Glucocorticoids act through binding to the cytosolic glucocorticoid receptor, a member of the superfamily of ligand regulated nuclear receptors [7]. Human glucocorticoid receptor, cloned in 1984, is comprised of 777 amino acids and contains a DNA binding domain (DBD), a ligand binding domain (LBD), and ԏ1 and ԏ2 activation domains. Glucocorticoid receptor exists predominantly in the cytoplasm as an inactive complex with other proteins, including hsp90 as well as immunophilins p59 and calrecticulin [8-9]. In the cytoplasm, binging of glucocorticoid to the LBD of the glucocorticoid receptor causes receptor conformational change, leading to dissociation of the receptor-protein complex and translocation of glucocorticoid receptor into the nucleus, where it binds to glucocorticoid response elements (GRE) in the promoter region of targeted genes and regulates gene expression, either transactivation or transregression. Notably, lipocortin I or annexin I, an inhibitor of arachidonic acid release from plasma membrane, is up-regulated by glucocorticoid [10]. Glucocorticoids can act through delayed, genomic mechanisms (as described above) as well as rapid, non-genomic effects [11].
In addition to systemic regulation, glucocorticoid effects are also regulated locally in tissues by a "pre-receptor" regulatory mechanism involving 11β-hydroxysteroid dehydrogenase type I (11βHSD1) and 11βHSD2 [12]. 11βHSD1 produces active glucocorticoids from inactive metabolites, while 11βHSD2 converts glucocorticoids to their inactive ketoforms. 11βHSD1 is expressed in key glucocorticoid target tissues such as liver and adipose tissue, while 11βHSD2 is expressed predominantly in mineralocorticoid responsive tissues such as the kidney, salivary gland and colon [13-16]. In addition to the activation of glucocorticoid receptors, glucocorticoids also possess the potential to activate the mineralocorticoid receptor. The concentration of glucocorticoids in the circulation is ~ 10-6 M, whereas the concentration of aldosterone in the circulation is ~ 10-9 M [17]. Therefore, the physiological function of 11βHSD2 is to protect mineralocorticoid receptors from activation by cortisol (human) or corticosterone (CS, rodents), thus providing mineralocorticoid receptor selectivity to aldosterone [12]. The syndrome of apparent mineralocorticoid excess (AME) is caused by mutation in exon 5 of 11βHSD2, which leads to a premature stop site at codon 374 instead of a normal arginine (R374), with the deletion of 32 amino acids from the C-terminus. AME, an autosomal recessive, is characterized by hypertension, hypokalaemia and suppression of the renin-angiotensin-aldosterone axis due to cortisol activation of mineralocorticoid receptors as a result of deficiency of 11βHSD2 activity [18]. AME is an example of human hypertension arising from a single gene defect.
11βHSD2 acts as a procarcinogenic agent through attenuation of glucocorticoid actions
Glucocorticoids have been used for decades in the treatment of hematologic malignancies [3-5]. In recent years, glucocorticoids have also been used in the treatment of solid tumors [19]. Glucocorticoids are extensively used in combination chemotherapy of advanced prostate cancer [20]. Dexamethasone has been used to inhibit colorectal cancer hepatic metastasis [21]. In experimental studies, glucocorticoids have been shown to effectively prevent the development of lung tumors in A/J mice as well as the development of skin tumors [22-24]. Liposomal prednisolone (glucocorticoid) phosphate inhibited tumor growth dose-dependently, with 80% to 90% tumor growth inhibition of subcutaneous B16.F10 melanoma and C26 colon carcinoma murine tumor models at 20 mg/kg by single or weekly doses [25]. Dexamethasone inhibited prostate tumorigenesis through inhibition of tumor-associated angiogenesis by decreasing VEGF through activation of glucocorticoid receptors [26-27].
Recently, 11βHSD2 has been proposed to be a pro-proliferative agent due to its ability to attenuate the anti-proliferative effects of glucocorticoids. The most conclusive evidence for the role of 11βHSD2 in tumorigenesis comes from the analysis of 11βHSD isoenzyme expression in a large cohort of human pituitary tumors [28-30]. 11βHSD1 expression decreases, while 11&βHSD2 expression dramatically increases in pituitary tumors compared with normal tissues. Inhibition of 11βHSD2 activity suppresses pituitary cell proliferation and augments glucocorticoids anti-proliferative effects. 11βHSD2 expression increases in human adrenal cortical carcinoma/adenoma, breast cancers, and neoplastic cell lines [31-36]. In cultured cell lines, overexpression of 11βHSD2 stimulates cell proliferation, while inhibition of 11βHSD2 activity suppresses cell proliferation [32-35].
Cyclooxygenase-2 and colorectal tumorigenesis
In 1934, Nobel laureate (1970, Medicine or Physiology) Ulf von Euler found that the injection of extracts of sheep vesicular gland dramatically decreased blood pressure. Extracts from human seminal fluid had a similar effect. Although von Euler named the substance prostaglandin (PG) due to the mistaken belief that it originated in the prostate gland, we did find that cyclooxygenase-2, the major enzyme to mediate prostaglandin production, is constitutively expressed in distal vas deferens of the prostate and its expression is many fold greater than in any other organs of the body and is regulated by androgens [37]. In the 1950s, Bergstrom and his associates purified two important prostaglandins, PGE and PGF, and identified their chemical structure. Bengt I Samuelsson elucidated arachidonic acid and prostaglandin metabolism and discovered endoperoxides, thromboxanes and leukotrienes. Aspirin is the most frequently used drug all over the world. Sir John Vane discovered prostacyclin and found that aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) act through inhibition of prostaglandin biosynthesis. For their contributions to the crucial breakthrough in prostaglandin research, Sune Karl Bergstrom, Bengt I Samuelsson and John Vane shared the Nobel Prize in Medicine and Physiology in 1982. Until the late 1980s, only one isoform of COX had been identified (now recognized as COX-1). Needleman et al. first proposed the existence of an inducible, glucocorticoid-suppressive cyclooxygenase activity, which was then identified by two independent groups, and is now called COX-2 [38-40]. COX-1 is responsible for 'housekeeping' prostaglandin biosynthesis and is constitutively expressed in most tissues, while COX-2 is induced by growth factors and pro-inflammatory cytokines, and exquisitely suppressed by glucocorticoids.
During the development of CRC, prostaglandin production, particularly the E series of prostaglandins, is significantly increased in malignant tissues. Decreased polyp multiplicity and regression of polyps with long-term use of sulindac in patients with familial adenomatous polyposis (FAP) provided the first insights into the potential chemopreventive actions of NSAIDs on CRC [41-42]. Multiple studies have shown a 40-50% reduction in CRC in individuals taking NSAIDs regularly either in the context of sporadic CRC or in FAP patients [43-59]. A recent retrospective cohort study indicates that aspirin reduces cancer risk in patients with high levels of COX-2 in the tumors, and regular aspirin use after the diagnosis of colorectal cancer improves overall survival, particularly in patients with high levels of COX-2 in the tumors [60-61]. In a most recent report, regular use of aspirin is reported to be associated with longer survival among CRC patients with PIK3CA mutation [62]. NSAIDs reduce the relative risk of CRC primarily due to their ability to inhibit prostaglandin production [63]. However, the chemopreventive and therapeutic application of NSAIDs to CRC is limited due to adverse side effects (gastric ulceration and perforation) [64]. Reports estimate 10,000 to 20,000 deaths and 100,000 hospitalizations per year in the United States related to traditional NSAIDs [65]. The anti-inflammatory properties of NSAIDs are attributed to the inhibition of COX-2, while the gastrointestinal side effects of NSAIDs are attributed to the inhibition of COX-1 [63-66].
COX-2 expression increases in Apc+/min (multiple intestinal neoplasms) and ApcΔ716 (adenomatous polyposis coliΔ716) mouse adenomas, in azoxymethane-induced colonic cancer, and in human colorectal adenomas and cancers [67-68]. COX-2 also plays an important role in CRC metastasis [69-72]. Hepatic metastases develop more frequently when primary CRC expresses high COX-2 levels. COX-2 expression increases not only in the primary CRC, but to even higher levels in hepatic metastases [69]. Deletion of the COX-2 gene suppresses adenoma development in ApcΔ716 mice and Apc+/min mice [73-74]. Therefore, COX-2 is involved in colorectal tumorigenesis at multiple stages. COX-2-derived PGE2 promotes tumor growth and metastasis through stimulation of cell proliferation, cell migration, cell invasion, angiogenesis, and immunosuppression [70-72-75-80].
Selective COX-2 inhibitors were developed based on the hypothesis that COX-2 was the major source of prostaglandins that mediate inflammation while COX-1 was the source of cytoprotective prostaglandins in the gastric epithelium [81]. COX-2 inhibitors that reached the market (such as celecoxib) have shown similar efficacy to the traditional NSAIDs in the treatment of acute and chronic inflammatory conditions, but with fewer gastrointestinal side effects [82-83]. Selective COX-2 inhibitors have been shown to be effective in rodent models of colonic carcinogenesis [67-84-89]. Clinically, celecoxib, a selective COX-2 inhibitor, significantly reduced the number and burden of colorectal polyps in FAP patients [90-91]. However, long-term use of high doses of selective COX-2 inhibitor is currently not recommended because of the unacceptable cardiovascular side effects in certain patients, especially those with a history of atherosclerotic heart disease [92]. Possibly due to inhibition of endothelial cell-derived COX-2 activity, with selective inhibition of PGI2 production, leaving COX-1-mediated production of thromboxane A2 and subsequent platelet aggregation unchecked [81, 93-96]. Long-term use of COX-2 inhibitors also increases blood pressure [97-99].
Inhibition of 11βHSD2 activity suppresses colorectal tumorigenesis through enhancing glucocorticoid-mediated inhibition of COX-2 pathway
Since its discovery, COX-2 has been found to be suppressed by glucocorticoids via glucocorticoid receptors in all cell types and tissues. Activation of glucocorticoid receptors inhibits COX-2 expression primarily through three mechanisms [7]: 1. Transcriptional inhibition: Activation of either AP-1 or NF-κB leads to increased COX-2 transcription. Glucocorticoid receptors directly inhibit COX-2 expression by interaction with AP-1 and NF-κB; 2. Indirect transcriptional inhibition: Glucocorticoids induce IκBα, which directly inhibits NF-κB activity. Glucocorticoids also induce MAPK phosphatase-1 (MKP-1), which inhibits p38, ERK and JNK activities, leading to inhibition of NF-κB and AP-1. Glucocorticoids also induce glucocorticoid-induced leucine zipper, which directly inhibits transcription of both AP-1 and NF-κB; 3. Post-transcriptional inhibition: Sustained p38 activity is required for the stabilization of COX-2 mRNA. MKP-1 induced by glucocorticoids inactivates p38, leading to destabilization of COX-2 mRNA.
Recent studies have demonstrated a clear molecular link between COX-2-derived PGE2 activation of prostaglandin E receptor type 2 (EP2) and colorectal cancer progression [100]. Glucocorticoids and NSAIDs inhibit prostaglandin biosynthesis through different mechanisms. NSAIDs inhibit prostaglandin biosynthesis by non-competitive inhibition of both COX-1 and COX-2 enzymatic activity. Glucocorticoids are the most potent, endogenous, specific COX-2 inhibitors. Glucocorticoids suppress prostaglandin production through inhibiting cytosolic phospholipase A2 activity and suppressing COX-2 and mPGES-1 expression [7,101,102]. The concentration of glucocorticoids in the circulation is ~ 10-6 M [103]. In cultured cells, COX-2 expression is inhibited by glucocorticoids in the range of 10-10-10-7 M. For example, in cultured mouse medullary interstitial cells, which express high levels of COX-2 but do not express detectable 11βHSD2, COX-2 expression is inhibited by 10-9 M CS [103]. We found that there is constitutive COX-2 expression in epithelial cells in the macula densa and adjacent cortical thick ascending limbs in kidney cortex [104,105]. Renal cortical COX-2 expression increases after adrenalectomy (glucocorticoid and mineralocorticoid deficiency). Glucocorticoid replacement with corticosterone reverses the cortical COX-2 elevation seen in adrenalectomized rats. Inhibition of the glucocorticoid receptors with RU486 increases cortical COX-2 expression. Inhibition of 11βHSD2 activity with glycyrrhizic acid leads to suppression of renal cortical COX-2 expression, which is reversed by the glucocorticoid receptor antagonist RU486 [99]. These results indicate that 11βHSD2 inhibition may mimic glucocorticoids to suppress COX-2 expression by attenuating glucocorticoid inactivation, thereby increasing intracellular active glucocorticoids.
11βHSD2 activity is higher in the colon than in the small intestine in the rat and it is primarily localized to intestinal epithelia [106]. In intestinal tumors, βHSD2 was expressed in epithelial and stromal cells [36,107,108]. Interestingly, the incidence of intestinal tumors is always higher in the colon than in the small intestine. The mechanism underlying increase in COX-2 expression in colorectal cancer is not completely understood. We recently found that 11βHSD2 mRNA levels were significantly higher in both human colonic small and large adenomas comparing to adjacent normal colon tissues [101]. Immunohistochemical staining showed that increased 11βHSD2 expression was primarily localized to adenoma epithelia and co-localized with increased COX-2 expression. Both 11βHSD2 and COX-2 expression also increased in adenomas of Apc+/min mice. Mouse adenocarcinoma CT26 cells demonstrate significant tumorigenic activity and express high levels of COX-2, and both their proliferation in vitro and the size and number of CT26 tumors in vivo are sensitive to COX-2 inhibition [109]. CT26 cells also express 11βHSD2. Corticosterone treatment (the active glucocorticoid in rodents) leads to inhibition of CT26 cell COX-2 expression, which is enhanced by pharmacologic or genetic inhibition of 11βHSD2 activity but is attenuated by overexpression of 11βHSD2. Pharmacologic inhibition of 11βHSD2 activity with glycyrrhizic acid (GA) suppresses adenoma development and growth, associated with inhibition of adenoma COX-2 and mPGES-1 expression in Apc+/min mice. Knockdown of 11βHSD2 in CT26 cells leads to inhibition of CT26 tumor growth associated with inhibition of CT26 tumor COX-2 and mPGES-1 expression as well as PGE2 production. Pharmacologic inhibition of 11βHSD2 activity with GA increases active glucocorticoid levels but decreases inactive 11-keto-corticosterone levels in CT26 tumors as well as in tissues with high levels of 11βHSD2 activity such as kidney and colon. GA treatment leads to decreases in CT26 tumor incidence, inhibition of CT26 tumor growth, decreases in tumor phosphorylated cPLA2 (active form), COX-2 and mPGES-1 expression as well as tumor PGE2 levels; all of these parameter are reversed by treatment with RU486, an inhibitor of glucocorticoid receptors. Either genetic or pharmacologic inhibition of 11βHSD2 activity suppresses CT26 tumor metastasis. Human colon carcinoma HCA-7 cells constitutively express both 11βHSD2 and COX-2, and COX-2 activity is essential for the proliferation of HCA-7 cells [110]. On the other hand, human colon carcinoma HT-29 cells constitutively express similar levels of 11βHSD2 with minimal COX-2 expression, and COX-2 activity is not essential for HT-29 cell proliferation [111]. Inhibition of 11βHSD2 activity with glycyrrhizic acid has no effect on HT-29-derived tumor growth, but significantly inhibits HCA-7-derived tumor growth, which is associated with decreases in tumor COX-2 and mPGES-1 expression. Therefore, 11βHSD2 inhibition represents a novel approach for colorectal cancer chemoprevention and therapy by increasing tumor glucocorticoid activity, which in turn selectively blocks local COX-2 activity.
Other mechanisms by which 11βHSD2 inhibition may suppress colorectal tumorigenesis
In addition to inhibition of COX-2 pathway, increased intracellular glucocorticoids due to 11βHSD2 inhibition may suppress proliferation and induce apoptosis and differentiation through other mechanisms. The retinoblastoma protein (Rb), a tumor suppressor, inhibits cell proliferation through its interaction with the E2F family of transcription factors and the resultant regression of genes that are essential for DNA synthesis [112]. The inactivation of retinoblastoma protein is a prerequisite for cell proliferation. Inactivation of Rb is achieved through cyclin-dependent protein kinase-mediated phosphorylation during cell cycle progression. Glucocorticoids inactivate Rb through p53-mediated induction of p21waf1/cip1, an inhibitor of cyclin-dependent kinase [105105,113-116]. Glucocorticoids may also inhibit tumor growth through suppression of the mTOR signal pathway [117,118]. REDD1 (RTP801, an mTOR complex 1, mTORC1, repressor) is a novel stress-induced gene linked to regression of mTOR signaling and is induced by glucocorticoids [119-121]. Recently, we generated Apc+/min mice with selective deletion of 11βHSD2 in intestinal epithelial cells, and intestinal tumorigenesis is inhibited in these mice (our unpublished data). In adenomas from these mice, p53 and p21 levels increased, with concomitant decrease in phosphorylation of retinoblastoma protein. In addition, adenoma REDD1 increased, while the activity of mTOR signaling pathway was inhibited.
The lipoxygenase (LOX) pathway is another important pathway for metabolism of arachidonic acid. 5-LOX and its metabolite, leukotriene B4, have been reported to stimulate the proliferation of several human colon carcinoma cell lines [121-123]. 5-LOX increases in human colorectal tumors, and inhibition of the 5-LOX pathway suppresses the proliferation of human colon carcinoma cell lines and inhibits CRC development [124-127]. Dexamethasone has been reported to inhibit 5-LOX expression and its metabolite production [128-130].
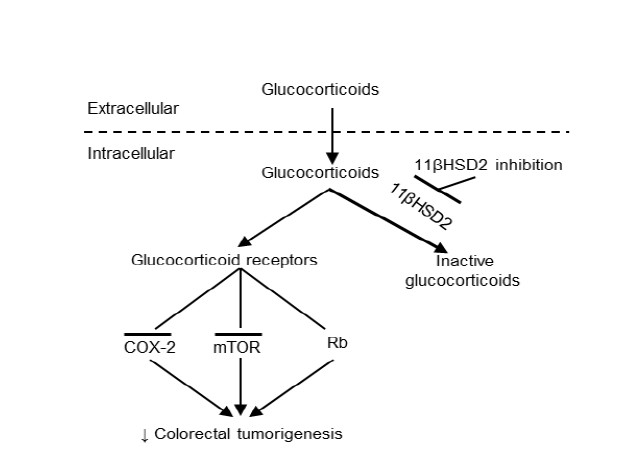
Figure 1: Proposed mechanism underlying epithelial cell 11ßHSD2 activity and colorectal tumorigenesis. In tumor epithelial cells, glucocorticoids are converted to inactive 11-keto-forms, reducing glucocorticoid receptor activity, while 11ßHSD2 inhibition leads to increased levels of epithelial intracellular active glucocorticoid. The susequent inhibition of the COX-2 pathway and induction of G1 cell cycle arrest through activation of tetinoblastoma protein (Rb, a tumor suppressor) and inhibition of the mTOR pathway lead to inhibition of colorectal tumorigenesis.
Conclusion and Future Perspective
We propose that inhibition of 11βHSD2 activity may provide a new target for chemoprevention and/or adjunctive therapy for colorectal cancers, particularly for patients with increased risk, such as familial adenomatous polyposis (FAP) patients, as a result of increased tumor intracellular active glucocorticoids, because of the following advantages (Figure 1): 1) Glucocorticoids selectively inhibit COX-2, but not COX-1, mediated prostaglandin production. Therefore, inhibition of 11βHSD2 activity has the beneficial effects of traditional NSAIDs to prevent and regress colorectal cancers without the gastrointestinal side effects associated with COX-1 inhibition; 2)Physiologic 11βHSD2 expression is most highly expressed incolon and kidney. Therefore, inhibition of 11βHSD2 activity is not expected to incur the cardiovascular risk posed by COX-2 inhibitors that suppress COX-2-derived PGI2 production in vascular endothelial cells; 3) Increased levels of intracellular active glucocorticoids are observed only in tissues with elevated 11βHSD2 expression. Inhibition of 11βHSD2 will not produce immunosuppression or other systemic side effects of conventional glucocorticoid therapy; 4) Increased tumor active glucocorticoids also inhibit tumor development and growth through induction of G1 cell cycle arrest and inhibition of the mTOR pathway; 5) Tissue prostaglandin levels are determined by both biosynthesis and catabolism. A key enzyme in prostaglandin catabolism, 15-hydroxyprostaglandin has been reported to be decreased in Apc+/min mouse intestinal adenomas and in human colorectal cancer [131], and glucocorticoids have been shown to induce 15-hydroxyprostaglandin in A549 human lung adenocarcinoma cells [132]. Therefore, it is possible that increased intracellular active endogenous glucocorticoids may inhibit colorectal tumorigenesis not only by inhibiting prostaglandin synthesis, but also by enhancing prostaglandin degradation; and finally 6)Glucocorticoid also inhibit the activity of cPLA2, preventingpotential shunting of arachidonic acid metabolism to other procarcinogenic metabolizing pathway such as 5-lipoxygenase lipoxygenase pathway [128-130,133]. In addition, glucocorticoids inhibit both COX-2 and 5-LOX.Glycyrrhizic acid and its analogs are excellent prototypes for 11βHSD2 inhibitors. It is a natural compound contained in licorice, a natural botanical antiinflammatory agent and a powerful 11βHSD2 inhibitor.
Long-term excessive ingestion of licorice has been reported to induce hypokalemia and elevation of blood pressure in a subset of people [134]. Although we did not see these side effects in our experimental animals with glycyrrhizic acid treatment, we did observe that the concentrations of glycyrrhizic acid used in our studies increased levels of active glucocorticoid levels in the kidney [101]. Therefore, it is likely that if comparable doses of glycyrrhizic acid were used in humans, some percentage of patients would develop hypertension and/or hypokalemia, requiring treatment with the potassium-sparing diuretic amiloride. However, the need to monitor for these potential side effects should not necessarily preclude the use of 11βHSD2 inhibition as a strategy to inhibit colorectal tumor COX-2 expression and growth. In addition, development of locally acting enteric 11βHSD2 inhibitors that are not systemically absorbed would be a potential therapeutic means to prevent colorectal tumorigenesis [61].
Acknowledgements
This work was supported by grants from the National Institutes of Health (CA122620, DK38226, DK51265, DK62794) and by the Vanderbilt Ò Brien Center (DK79341).References
- Parkin DM (2001) Global cancer statistics in the year 2000. The Lancet Oncol 2: 533-543.
- Clark AR, Lasa M (2003) Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr Opin Pharmacol 3: 404-411.
- Frankfurt O, Rosen ST (2004) Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 16: 553-563.
- Almawi WY, Abou Jaoude MM, Li XC (2002) Transcriptional and post-transcriptional mechanisms of glucocorticoid antiproliferative effects. Hematol Oncol 20: 17-32.
- Sionov RV, Spokoini R, Kfir-Erenfeld S, Cohen O, Yefenof E (2008) Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Adv Cancer Res 101: 127-248.
- Stewart PM, Prescott SM (2009) Can licorice lick colon cancer? J Clin Invest 119: 760-763.
- Newton R (2000) Molecular mechanisms of glucocorticoid action: what is important? Thorax 55: 603-613.
- Beato M, Sanchez-Pacheco A (1996) Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev 17: 587-609.
- Beato M, Candau R, Chavez S, Mows C, Truss M (1996) Interaction of steroid hormone receptors with transcription factors involves chromatin remodelling. J Steroid Biochem Mol Biol 56: 47-59.
- McKanna JA, Zhang MZ (1997) Immunohistochemical localization of lipocortin 1 in rat brain is sensitive to pH, freezing, and dehydration. J Histochem Cytochem 45: 527-538.
- Groeneweg FL, Karst H, de Kloet ER Joels M (2011) Rapid non-genomic effects of corticosteroids and their role in the central stress response. J Endocrinol 209: 153-167.
- Funder, JW, Pearce PT, Smith R, Smith AI (1988) Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242: 583-585.
- Masuzaki H (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166-2170.
- Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, et al. (1999) Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest 103: 683-689.
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, et al. (1997) 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 94: 14924-14929.
- Moore XL, Hoong I, Cole TJ (2000) Expression of the 11beta-hydroxysteroid dehydrogenase 2 gene in the mouse. Kidney Int 57: 1307-1312.
- Zhang MZ, Wang SW, Cheng H, Zhang Y, McKanna JA, et al. (2003) Regulation of renal cortical cyclooxygenase-2 in young rats. Am J Physiol Renal Physiol 285: F881-888.
- Stewart PM, Krozowski ZS, Gupta A, Milford DV, Howie AJ, et al. (1996) Hypertension in the syndrome of apparent mineralocorticoid excess due to mutation of the 11 beta-hydroxysteroid dehydrogenase type 2 gene. Lancet 347: 88-91.
- Keith BD (2008) Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 8: 84.
- Yemelyanov A, Czwornog J, Chebotaev D, Karseladze A, Kulevitch E, et al. (2007) Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene 26: 1885-1896.
- Dizon DS, Kemeny NE (2002) Intrahepatic arterial infusion of chemotherapy: clinical results. Semin Oncol 29: 126-135.
- Yao R, Wang Y, Lemon WJ, Lubet RA, You M (2004) Budesonide exerts its chemopreventive efficacy during mouse lung tumorigenesis by modulating gene expressions. Oncogene 23: 7746-7752.
- Chebotaev D, Yemelyanov A, Zhu L, Lavker RM, Budunova I (2007) The tumor suppressor effect of the glucocorticoid receptor in skin is mediated via its effect on follicular epithelial stem cells. Oncogene 26: 3060-3068.
- Budunova IV, Kowalczyk D, Pérez P, Yao YJ, Jorcano JL, et al. (2003) Glucocorticoid receptor functions as a potent suppressor of mouse skin carcinogenesis. Oncogene 22: 3279-3287.
- Schiffelers RM, Metselaar JM, Fens MH, Janssen AP, Molema G, et al. (2005) Liposome-encapsulated prednisolone phosphate inhibits growth of established tumors in mice. Neoplasia 7: 118-127.
- Nishimura K, Nonomura N, Satoh E, Harada Y, Nakayama M, et al. (2001) Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J Natl Cancer Inst 93, 1739-1746.
- Yano A, Fujii Y, Iwai A, Kageyama Y, Kihara K (2006) Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clin Cancer Res 12: 3003-3009.
- Rabbitt EH, Ayuk J, Boelaert K, Sheppard MC, Hewison M, et al. (2003) Abnormal expression of 11 beta-hydroxysteroid dehydrogenase type 2 in human pituitary adenomas: a prereceptor determinant of pituitary cell proliferation. Oncogene 22: 1663-1667.
- Rabbitt EH, Gittoes NJ, Stewart PM, Hewison M (2003) 11beta-hydroxysteroid dehydrogenases, cell proliferation and malignancy. J Steroid Biochem Mol Biol 85: 415-421.
- Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM, et al. (2002) Prereceptor regulation of glucocorticoid action by 11beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J 16: 36-44.
- Coulter CL, Smith RE, Stowasser M, Sasano H, Krozowski ZS, et al. (1999) Expression of 11beta-hydroxysteroid dehydrogenase type 2 (11betaHSD-2) in the developing human adrenal gland and human adrenal cortical carcinoma and adenoma. Mol Cell Endocrinol 154: 71-77.
- Hundertmark S, Buhler H, Rudolf M, Weitzel HK, Ragosch V (1997) Inhibition of 11 beta-hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75-1 breast cancer cells. J Endocrinol 155: 171-180.
- Lipka C, Mankertz J, Fromm M, Lübbert H, Bühler H, et al. (2004) Impairment of the antiproliferative effect of glucocorticosteroids by 11beta-hydroxysteroid dehydrogenase type 2 overexpression in MCF-7 breast-cancer cells. Horm Metab Res 36: 437-444.
- Koyama K, Krozowski Z (2001) Modulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity in Ishikawa cells is associated with changes in cellular proliferation. Mol Cell Endocrinol 183: 165-170.
- Koyama K, Myles K, Smith R, Krozowski Z (2001) Expression of the 11beta-hydroxysteroid dehydrogenase type II enzyme in breast tumors and modulation of activity and cell growth in PMC42 cells. J Steroid Biochem Mol Biol 76: 153-159.
- Takahashi K, Sasano H, Fukushima K, Hirasawa G, Miura H, et al. (1998) 11 beta-hydroxysteroid dehydrogenase type II in human colon: a new marker of fetal development and differentiation in neoplasms. Anticancer Res 18: 3381-3388.
- McKanna JA, Zhang MZ, Wang JL, Cheng H, Harris RC (1998) Constitutive expression of cyclooxygenase-2 in rat vas deferens. Am J Physiol 275, R227-R233.
- Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P (1990) The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem 265: 16737-16740.
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR (1991) TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 266: 12866-12872.
- O'Banion MK, Sadowski HB, Winn V, Young DA (1991) A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J Biol Chem 266: 23261-23267.
- Waddell WR, Ganser GF, Cerise EJ, Loughry RW (1989) Sulindac for polyposis of the colon. Am J Surg 157: 175-179.
- Waddell WR, Loughry RW (1983) Sulindac for polyposis of the colon. J Surg Oncol 24: 83-87.
- Kune GA, Kune S, Watson LF (1988) Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 48: 4399-4404.
- Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW (1993) Aspirin use and risk of fatal cancer. Cancer Res 53: 1322-1327.
- Thun MJ, Namboodiri MM, Heath CW (1991) Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 325: 1593-1596.
- Bansal P, Sonnenberg A (1996) Risk factors of colorectal cancer in inflammatory bowel disease. Am J Gastroenterol 91: 44-48.
- Gann PH, Manson JE, Glynn RJ, Buring JE, Hennekens CH (1993) Low-dose aspirin and incidence of colorectal tumors in a randomized trial. J Natl Cancer Inst 85: 1220-1224.
- Giovannucci E, Egan KM, Hunter DJ, Stampfer MJ, Colditz GA, et al. (1995) Aspirin and the risk of colorectal cancer in women. N Engl J Med 333: 609-614.
- Giovannucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A,et al. (1994) Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Intern Med 121: 241-246.
- Greenberg ER, Baron JA, Freeman DH, Mandel JS, Haile R (1993) Reduced risk of large-bowel adenomas among aspirin users. The Polyp Prevention Study Group. J Natl Cancer Inst 85: 912-916.
- Muscat JE, Stellman SD, Wynder EL (1994) Nonsteroidal antiinflammatory drugs and colorectal cancer. Cancer 74: 1847-1854.
- Reeves MJ, Newcomb PA, Trentham-Dietz A, Storer BE, Remington PL (1996) Nonsteroidal anti-inflammatory drug use and protection against colorectal cancer in women. Cancer Epidemiol Biomarkers Prev 5: 955-960.
- Suh O, Mettlin C, Petrelli NJ (1993) Aspirin use, cancer, and polyps of the large bowel. Cancer 72: 1171-1177.
- Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, et al. (2003) A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 348: 891-899.
- Coogan PF, Rosenberg L, Louik C, Zauber AG, Stolley PD, et al. (2000) NSAIDs and risk of colorectal cancer according to presence or absence of family history of the disease. Cancer Causes Control 11: 249-255.
- Freedman AN, Michalek AM, Weiss HA, Zhang ZF, Marshall JR, et al. (1998) Aspirin use and p53 expression in colorectal cancer. Cancer Detect Prev 22: 213-218.
- Rosenberg L, Louik C, Shapiro S (1998) Nonsteroidal antiinflammatory drug use and reduced risk of large bowel carcinoma. Cancer 82: 2326-2333.
- Smalley WE, DuBois RN (1997) Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol 39: 1-20.
- Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, et al. (2003) A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med 348: 883-890.
- Chan AT, Ogino S, Fuchs CS (2007) Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med 356: 2131-2142.
- Chan AT, Ogino S, Fuchs CS (2009) Aspirin use and survival after diagnosis of colorectal cancer. Jama 302: 649-658.
- Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, et al. (2012) Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 367: 1596-1606.
- Brown JR, DuBois RN (2005) COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol 23: 2840-2855.
- Fries J (1996) Toward an understanding of NSAID-related adverse events: the contribution of longitudinal data. Scand J Rheumatol Suppl 102: 3-8.
- Miller JL (1999) Decisions loom on selective COX-2 inhibitors. Am J Health Syst Pharm 56: 106-107.
- Gupta S, Adhami VM, Subbarayan M, MacLennan GT, Lewin JS, et al. (2004) Suppression of Prostate Carcinogenesis by Dietary Supplementation of Celecoxib in Transgenic Adenocarcinoma of the Mouse Prostate Model. Cancer Res 64: 3334-3343.
- Oshima M, Murai N, Kargman S, Arguello M, Luk P, et al. (2001) Chemoprevention of intestinal polyposis in the Apcdelta716 mouse by rofecoxib, a specific cyclooxygenase-2 inhibitor. Cancer Res 61: 1733-1740.
- Williams CS, Luongo C, Radhika A, Zhang T, Lamps LW, et al. (1996) Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology 111: 1134-1140.
- Chen WS, Wei SJ, Liu JM, Hsiao M, Kou-Lin J, et al. (2001) Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer 91: 894-899.
- Kakiuchi Y, Tsuji S, Tsujii M, Murata H, Kawai N, et al. (2002) Cyclooxygenase-2 activity altered the cell-surface carbohydrate antigens on colon cancer cells and enhanced liver metastasis. Cancer Res 62: 1567-1572.
- Soumaoro LT, Uetake H, Higuchi T, Takagi Y, Enomoto M, et al. (2004) Cyclooxygenase-2 expression: a significant prognostic indicator for patients with colorectal cancer. Clin Cancer Res 10: 8465-8471.
- Tsujii M, Kawano S, DuBois RN (1997) Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA 94: 3336-3340.
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, et al. (1996) Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87: 803-809.
- Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, et al. (2000) Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res 60: 4705-4708.
- Holla VR, Wang D, Brown JR, Mann JR, Katkuri S, et al. (2005) Prostaglandin E2 regulates the complement inhibitor CD55/decay-accelerating factor in colorectal cancer. J Biol Chem 280: 476-483.
- Kundu N, Walser TC, Ma X, Fulton AM (2005) Cyclooxygenase inhibitors modulate NK activities that control metastatic disease. Cancer Immunol Immunother 54: 981-987.
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, et al. (1998) Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 93: 705-716.
- Yao M, Lam EC, Kelly CR, Zhou W, Wolfe MM (2004) Cyclooxygenase-2 selective inhibition with NS-398 suppresses proliferation and invasiveness and delays liver metastasis in colorectal cancer. Br J Cancer 90: 712-719.
- Rao CV, Reddy BS (2004) NSAIDs and chemoprevention. Curr Cancer Drug Targets 4: 29-42.
- Wallace JM (2002) Nutritional and botanical modulation of the inflammatory cascade--eicosanoids, cyclooxygenases, and lipoxygenases--as an adjunct in cancer therapy. Integr Cancer Ther 1: 7-37.
- FitzGerald GA (2003) COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov 2: 879-890.
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, et al. (2000) Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med 343: 1520-1528.
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, et al. (2000) Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. Jama 284: 1247-1255.
- Goldman AP, Williams CS, Sheng H, Lamps LW, Williams VP, et al. (1998) Meloxicam inhibits the growth of colorectal cancer cells. Carcinogenesis 19: 2195-2199.
- Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA (2000) The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res 60: 5040-5044.
- Kawamori T, Rao CV, Seibert K Reddy BS (1998) Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 58: 409-412.
- Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, et al. (2000) Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res 60, 293-297.
- Sawaoka H, Tsuji S, Tsujii M, Gunawan ES, Sasaki Y, et al. (1999) Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab Invest 79: 1469-1477.
- Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN (1998) Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res 58: 362-366.
- Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, et al. (2002) A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 50: 857-860.
- Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, et al. (2000) The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342: 1946-1952.
- Cuzick J, Otto F, Baron JA, Brown PH, Burn J, et al. (2009) Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol 10: 501-507.
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, et al. (2005) Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352: 1092-1102.
- Nussmeier NA, Whelton AA, Brown MT, Joshi GP, Langford RM, et al. (2006) Safety and efficacy of the cyclooxygenase-2 inhibitors parecoxib and valdecoxib after noncardiac surgery. Anesthesiology 104: 518-526.
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, et al. (2005) Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 352: 1081-1091.
- Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, et al. (2005) Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 352: 1071-1080.
- Francois H, Coffman TM (2004) Prostanoids and blood pressure: which way is up? J Clin Invest 114: 757-759.
- Solomon DH, Schneeweiss S, Levin R, Avorn J (2004) Relationship between COX-2 specific inhibitors and hypertension. Hypertension 44: 140-145.
- Yao B, Harris RC, Zhang MZ (2005) Interactions between 11beta-hydroxysteroid dehydrogenase and COX-2 in kidney. Am J Physiol Regul Integr Comp Physiol 288: R1767-R1773.
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS (2005) Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310: 1504-1510.
- Zhang MZ, Xu J, Yao B, Yin H, Cai Q, et al. (2009) Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest 119: 876-885.
- Stichtenoth DO, Thorén S, Bian H, Peters-Golden M, Jakobsson PJ, et al. (2001) Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol 167: 469-474.
- Zhang MZ, Hao CM, Breyer MD, Harris RC, McKanna JA (2002) Mineralocorticoid regulation of cyclooxygenase-2 expression in rat renal medulla. Am J Physiol Renal Physiol 283: F509-F516.
- Zhang MZ, Wang JL, Cheng HF, Harris RC, McKanna, JA (1997) Cyclooxygenase-2 in rat nephron development. Am J Physiol 273: F994-1002.
- Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, et al. (1994) Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest 94: 2504-2510.
- Smith RE, Maguire JA, Stein-Oakley AN, Sasano H, Takahashi K, et al. (1996) Localization of 11 beta-hydroxysteroid dehydrogenase type II in human epithelial tissues. J Clin Endocrinol Metab 81: 3244-3248.
- Whorwood CB, Ricketts ML, Stewart PM (1994) Epithelial cell localization of type 2 11 beta-hydroxysteroid dehydrogenase in rat and human colon. Endocrinology 135: 2533-2541.
- Zbankova S, Bryndova J, Kment M, Pacha J (2004) Expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in colorectal cancer. Cancer Lett 210: 95-100.
- Pozzi A, Yan X, Macias-Perez I, Wei S, Hata AN, et al. (2004) Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem 279: 29797-29804.
- Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, et al. (1997) Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 99: 2254-2259.
- Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S (2002) Selective cyclooxygenase-2 inhibitors show a differential ability to inhibit proliferation and induce apoptosis of colon adenocarcinoma cells. FEBS Lett 531: 278-284.
- Chau BN, Wang JY (2003) Coordinated regulation of life and death by RB. Nat Rev Cancer 3: 130-138.
- Cram EJ, Ramos RA, Wang EC, Cha HH, Nishio Y, et al. (1998) Role of the CCAAT/enhancer binding protein-alpha transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J Biol Chem 273: 2008-2014.
- Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, et al. (1998) Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem 273: 1998-2007.
- Reil TD, Kashyap VS, Sarkar R, Freishlag J, Gelabert HA (2000) Dexamethasone inhibits the phosphorylation of retinoblastoma protein in the suppression of human vascular smooth muscle cell proliferation. J Surg Res 92: 108-113.
- Addeo R, Casale F, Caraglia M, D'Angelo V, Crisci S, et al. (2004) Glucocorticoids induce G1 arrest of lymphoblastic cells through retinoblastoma protein Rb1 dephosphorylation in childhood acute lymphoblastic leukemia in vivo. Cancer Biol Ther 3: 470-476.
- Polman JA, Hunter RG, Speksnijder N, van den Oever JM, Korobko OB, et al. (2012) Glucocorticoids Modulate the mTOR Pathway in the Hippocampus: Differential Effects Depending on Stress History. Endocrinology 153: 4317-4327.
- Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, et al. (2011) Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13: 170-182.
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR (2006) Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281: 39128-39134.
- Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, et al. (2011) Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem 286: 30181-30189.
- Kumari R, Willing LB, Jefferson LS, Simpson IA, Kimball SR (2011) REDD1 (regulated in development and DNA damage response 1) expression in skeletal muscle as a surrogate biomarker of the efficiency of glucocorticoid receptor blockade. Biochem Biophys Res Commun 412: 644-647.
- Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, et al. (1995) Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim Biophys Acta 1258: 215-223.
- Bortuzzo C, Hanif R, Kashfi K, Staiano-Coico L, Shiff SJ, et al. (1996) The effect of leukotrienes B and selected HETEs on the proliferation of colon cancer cells. Biochim Biophys Acta 1300: 240-246.
- Ohd JF, Nielsen CK, Campbell J, Landberg G, Löfberg H, et al. (2003) Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology 124: 57-70.
- Barresi V, Grosso M, Vitarelli E, Tuccari G, Barresi G (2007) 5-Lipoxygenase is coexpressed with Cox-2 in sporadic colorectal cancer: a correlation with advanced stage. Dis Colon Rectum 50: 1576-1584.
- Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, et al. (2006) Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther 5: 2716-2726.
- Ye YN, Wu WK, Shin VY, Bruce IC, Wong BC, et al. (2005) Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 26: 827-834.
- Jame AJ, Lackie PM, Cazaly AM, Sayers I, Penrose JF, et al. (2007) Human bronchial epithelial cells express an active and inducible biosynthetic pathway for leukotrienes B4 and C4. Clin Exp Allergy 37: 880-892.
- Peters-Golden M, Sampson AP (2003) Cysteinyl leukotriene interactions with other mediators and with glucocorticosteroids during airway inflammation. J Allergy Clin Immunol 111: S37-S48.
- Torosyan Y, Dobi A, Naga S, Mezhevaya K, Glasman M, et al. (2006) Distinct effects of annexin A7 and p53 on arachidonate lipoxygenation in prostate cancer cells involve 5-lipoxygenase transcription. Cancer Res 66: 9609-9616.
- Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, et al. (2005) 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem 280: 3217-3223.
- Tong M, Tai HH (2005) 15-Hydroxyprostaglandin dehydrogenase can be induced by dexamethasone and other glucocorticoids at the therapeutic level in A549 human lung adenocarcinoma cells. Arch Biochem Biophys 435: 50-55.
- Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, et al. (2006) Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther 5: 2716-2726.
- Stormer FC, Reistad R, Alexander J (1993) Glycyrrhizic acid in liquorice--evaluation of health hazard. Food Chem Toxicol 31: 303-312.