
Research Article
*Address for Correspondence: Claudia A. Chiriboga, MD, MPH, Division of Pediatric Neurology, Department of Neurology, College of Physicians and Surgeons, Columbia University Medical Center, Harkness Pavillion 552, 180 Fort Washington Avenue, New York, NY, 10032, USA, E-mail: cac3@columbia.edu
Citation: Chiriboga CA, Kuhn L, Wasserman GA. Neurobehavioral and Developmental Trajectories Associated with Level of Prenatal Cocaine Exposure. J Neurol Psychol. 2014;2(2): 12.
Copyright © 2014 Chiriboga CA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 2, Issue: 2
Submission: 13 September 2014 | Accepted: 20 November 2014 | Published: 25 November 2014
 Survival analyses: We used Kaplan-Meier survival analyses to assess changes in the neurological findings over time [60]. Failure was defined as the age at the time of resolution of hypertonia (global or in the legs) identified at an age of 6 months [48]. Censoring occurred at the end of the study (24-month visit) or at the child’s age at the last follow-up. We used Cox proportional hazard models to assess the rate of resolution by level of cocaine exposure controlling for significant variables [61]; the termination event was the age at which 6 month neurological findings disappeared.
Survival analyses: We used Kaplan-Meier survival analyses to assess changes in the neurological findings over time [60]. Failure was defined as the age at the time of resolution of hypertonia (global or in the legs) identified at an age of 6 months [48]. Censoring occurred at the end of the study (24-month visit) or at the child’s age at the last follow-up. We used Cox proportional hazard models to assess the rate of resolution by level of cocaine exposure controlling for significant variables [61]; the termination event was the age at which 6 month neurological findings disappeared.
 Psychomotor development index (PDI): Cocaine exposure was unrelated to 6-month PDI scores, with or without adjustment for maternal IQ, high school education, age > 30 yrs, birth weight, marijuana and tobacco exposure (F= 3.687; p= 0.002; R2 = 0.101) (P = 0.52; 0.6 PDI points; ΔR square 0.002) [48]. On the other hand, cocaine exposure was significantly associated with 12-month PDI scores with and without adjustment for maternal IQ, gestational age, birth weight, microcephaly, and marijuana exposure (F= 4.817; p= 0.000; R2 = 0.107) (p = 0.04; -2.773 PDI points; ΔR square 0.016). Prenatal cocaine exposure was related to 24-month Peabody Total Motor Scale in univariate analyses (p= 0.02), but associations did not persist after adjustment for significant factors (infant gender, maternal IQ, gestational age, and lead level; F= 4.913; p= 0.001; R2= 0.094) (Table 2). Analyses run with and without the inclusion of gestational age, microcephaly or birth weight did not alter the association between cocaine and motor outcome (PDI or Peabody score).
Psychomotor development index (PDI): Cocaine exposure was unrelated to 6-month PDI scores, with or without adjustment for maternal IQ, high school education, age > 30 yrs, birth weight, marijuana and tobacco exposure (F= 3.687; p= 0.002; R2 = 0.101) (P = 0.52; 0.6 PDI points; ΔR square 0.002) [48]. On the other hand, cocaine exposure was significantly associated with 12-month PDI scores with and without adjustment for maternal IQ, gestational age, birth weight, microcephaly, and marijuana exposure (F= 4.817; p= 0.000; R2 = 0.107) (p = 0.04; -2.773 PDI points; ΔR square 0.016). Prenatal cocaine exposure was related to 24-month Peabody Total Motor Scale in univariate analyses (p= 0.02), but associations did not persist after adjustment for significant factors (infant gender, maternal IQ, gestational age, and lead level; F= 4.913; p= 0.001; R2= 0.094) (Table 2). Analyses run with and without the inclusion of gestational age, microcephaly or birth weight did not alter the association between cocaine and motor outcome (PDI or Peabody score).
 Longitudinal analyses
Longitudinal analyses
 Motor development Z-scores: With regard to motor development, cocaine-exposed children had lower average motor development scores than cocaine-unexposed children but findings were not significant. Motor index scores did not decline across age. The motor Z-score, comprised of the z-score transformation of the 6 and 12 month PDI and the 24 month Peabody score, was unrelated to prenatal cocaine exposure in unadjusted analyses (p = 0.10), as well as in adjusted analyses (birth weight and gender) (p = 0.14). Age was not associated with motor scores. Analyses that excluded birth weight did not alter the association with cocaine and motor development.
Motor development Z-scores: With regard to motor development, cocaine-exposed children had lower average motor development scores than cocaine-unexposed children but findings were not significant. Motor index scores did not decline across age. The motor Z-score, comprised of the z-score transformation of the 6 and 12 month PDI and the 24 month Peabody score, was unrelated to prenatal cocaine exposure in unadjusted analyses (p = 0.10), as well as in adjusted analyses (birth weight and gender) (p = 0.14). Age was not associated with motor scores. Analyses that excluded birth weight did not alter the association with cocaine and motor development.

Neurobehavioral and Developmental Trajectories Associated with Level of Prenatal Cocaine Exposure
Claudia A. Chiriboga1,2,5*, Louise Kuhn2 and Gail A. Wasserman3,4
- 1Division of Pediatric Neurology, Department of Neurology, College of Physicians and Surgeons, Columbia University, New York, NY, USA
- 2Sergievsky Center, Columbia University, New York, NY, USA
- 3Division of Child and Adolescent Psychiatry, Department of Psychiatry, Columbia University, New York, NY, USA
- 4New York Psychiatric Institute, New York, NY, USA
- 5Harlem Hospital Center, New York, NY, USA
*Address for Correspondence: Claudia A. Chiriboga, MD, MPH, Division of Pediatric Neurology, Department of Neurology, College of Physicians and Surgeons, Columbia University Medical Center, Harkness Pavillion 552, 180 Fort Washington Avenue, New York, NY, 10032, USA, E-mail: cac3@columbia.edu
Citation: Chiriboga CA, Kuhn L, Wasserman GA. Neurobehavioral and Developmental Trajectories Associated with Level of Prenatal Cocaine Exposure. J Neurol Psychol. 2014;2(2): 12.
Copyright © 2014 Chiriboga CA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 2, Issue: 2
Submission: 13 September 2014 | Accepted: 20 November 2014 | Published: 25 November 2014
Abstract
Introduction: In experimental models, prenatal cocaine exposure has been found to perturb GABA and dopamine development. Clinically, abnormalities in tone, posture and state regulation are noted in early infancy but the evolution of these findings over time is not well described. The current study assesses the longitudinal effects of prenatal cocaine exposure in dose-dependent fashion on developmental & behavioral and neurological trajectories over the first 2 years of life.Methods: Three hundred and eighty infants, 113 cocaine-exposed, were enrolled at birth from an urban hospital from 2000 to 2004. Exposure was determined by maternal interview, segmental hair analyses (RIAH™) in all, and meconium and urine in a subset. Developmental, behavioral and neurological assessments were carried out blind to drug exposure at 6, 12 and 24 months of age in the 306 children who returned in follow-up. Mixed-effects linear modeling (developmental growth curve) assessed change in outcome over time.
Results: The mental developmental growth curve showed a negative slope (2.2 points) in adjusted analyses among cocaineexposed children over the first 2 years of life. (p=.04), while the slope of the motor development growth curve did not. Adjusting for microcephaly at 6 months diminished the strength of the association between cocaine exposure and mental developmental growth curve (effect modification). Cocaine exposure was marginally associated with behavioral outcomes in adjusted analyses. Total Behavior scores and Orientation/Engagement scores improved with age. At 1 year of age, prenatal cocaine exposure was significantly associated with lower motor development scores. High rates of hypertonia (global and diparesis) identified at the 6-month visit dropped dramatically in the first 2 years of life: cocaine-exposed children showed a more rapid rate of resolution of hypertonia than unexposed children, with hypertonia improving 2.2 times faster among those with heavy cocaine exposure.
Conclusion: We found differences in mental performance over the first 2 years of life associated with prenatal cocaine exposure that was mediated by microcephaly implying that cocaine exerts a sustained teratogenic effect on brain development. Early neurological (hypertonia) and behavioral findings associated with prenatal cocaine exposure improved over time. Hypertonia did not predict longterm development impairments. Conceivably, the transient nature of neurobehavioral manifestations reflects postnatally a tendency towards homeostasis of cocaine-related embryopathic perturbations of GABA and dopaminergic systems.
Keywords
Perinatal; Cocaine exposure; Behavior, Drug use; Hypertonia; Neurologic; Child developmentIntroduction
Whether cocaine has deleterious effects on the developing nervous system has been subject to debate. Over the last 2 decades, numerous controlled studies have focused on cognitive, developmental and behavioral differences associated with prenatal cocaine exposure with somewhat disparate results. Some have reported developmental findings in infants and toddlers that include cognitive [1-4], language [5-7] and motor delays [8-11], while another group found no specific cocaine effects across a range of abilities (development and language) [12]. Despite the developmental effects reported in young children, most studies in older children find no significant general or global cognitive effects associated with cocaine exposure [13-16], but instead report on more subtle effects affecting a narrow range of cognitive abilities (e.g. executive function [17,18] and perceptual reasoning [19]). Global language delays are reported associated with in utero cocaine exposure in children [5-7] and adolescents [20]. As with cognition, some groups report no abnormalities in language [21], whiles others report more subtle and narrow effects on language involving syntax and phonology [22].Prenatal cocaine exposure is associated with abnormal behaviors in offspring in both experimental [23,24] and clinical studies [25-39]. Exposed infants and young children are reported to be excessively irritable [25], exhibit dysregulation of arousal [26], and attention [26-28] and show problems with impulse control [29]. Older cocaine exposed children are noted to show higher rates of poor inhibitory control [30,31], impaired sustained visual attention [32], externalizing behaviors [33-38] including aggression [30], and increase in baseline arousal [38]. Other groups report no such abnormalities [39].
Unlike opiate exposure, prenatal exposure to cocaine does not exhibit a typical neonatal withdrawal syndrome; nonetheless, abnormalities of tone and posture, and state dysregulation are noted in prenatally exposed neonates [26,40-43]. Many of the behavioral and neurological findings are reported to linger into infancy but the evolution of these findings over time is not well described.
Only a handful of studies have prospectively assessed neurological function among cocaine-exposed children outside the newborn period [44-48] and even fewer have quantified prenatal cocaine exposure with the use of biomarkers [48,49]. Lewis et al. described higher rates of abnormal findings in neurological examination (either suspected or definitive) among cocaine-exposed toddlers, but the authors did not define further the nature or evolution of those neurological abnormalities [45]. In the present cohort of children we described higher rates of muscle tone abnormalities, particularly hypertonia (increase in extensor and flexor tone), associated with cocaine exposure in analyses that were carefully controlled for perinatal drug exposures at 6 months of age [48] but the implication of these findings, if any, for long term development is unknown.
Neurodevelopmental trajectories methods focus on the rate of change in outcome over time, i.e. slope of the developmental or behavioral trajectories for each child. This approach is ideally suited to assess the evolution of early neurobehavioral findings identified previously in our cohort. Herein we report on the developmental and behavioral trajectory, as well as the evolution of neurological signs, over the first two years of life in infants who enrolled at delivery in a longitudinal study. Prenatal exposure to cocaine and other drugs of abuse was determined at the time of birth with multiple biomarkers.
The current study was designed to assess the effect of cocaine exposure on developmental, behavioral, and neurological outcome with focus on changes over time. The primary hypothesis was that children with prenatal cocaine exposure would exhibit higher rates of developmental, behavioral, and neurological differences in dosedependent fashion compared to cocaine-unexposed children of comparable demographic background, after adjusting for potentially confounding factors. We postulated based on the results of our earlier studies [46] that hypertonia noted in infancy (6-month assessment) would improve over the first 2 years of life. We also sought to assess whether microcephaly, birth weight or hypertonia mediated any cocaine effects on development.
Methods
ProceduresStudy methods are described in detail elsewhere [48]. In brief, women were interviewed soon after delivery with a structured protocol that inquired about demographic information, obstetrical and medical history and drug use during and prior to the index pregnancy. Women were also administered an IQ test and a depression scale. Biological markers of drug exposure were obtained at the time of delivery (hair in all, meconium and urine in a subset). Women and their infants were invited at 6, 12, and 24 months of age for neurological, developmental and behavioral assessments. The biological mother was the primary caregiver in most cases (n = 296; 97%); 1 child was in the care of the father, the remainder in the care of a grandmother.
Participants
Beginning in May 2000 through November 2004 we prospectively enrolled mother-infant dyads from a municipal New York City hospital at the time of delivery as part of a prenatal drug exposure study. Informed consent was obtained at the time of enrollment. The study was approved by the Columbia Institutional Review Board. Maternal exclusionary criteria included non-English speaking, HIV infection, history of intravenous drug abuse and psychosis or bipolar disorder (as determined by chart review). Infants were excluded if <33 weeks gestational age or if there was a presence of major congenital malformation or birth asphyxia (5 minute Apgar ≤ 4).
Of women delivering at the hospital (n=3671), we approached 2,308: 675 were eligible and 1633 were ineligible primarily due to language constraints (non-English speaking). Of eligible women approached for enrollment (N=675), 277 refused. We, thus, enrolled 398 mother-infant dyads (59% of eligible women) into the study; of these, two were subsequently excluded due to medical factors (HIV and Trisomy 21) and 16 withdrew from the study. The final cohort thus comprises 380 mother-infant pairs. We excluded 10 opiateexposed children from controlled analyses.
Comparisons of women who refused enrollment with those enrolled revealed no differences in offspring gender, birth weight, birth length, or type of delivery (vaginal versus cesarean section) between groups (all p’s < 0.2). Women who refused enrollment, however, were older (25.8 yrs ± 6.6) than the women enrolled in the study (24.2 yrs ± 5.8, p= 0.007). Comparisons between those who returned for any visit (n=306) and those who never returned (n=74) revealed no significant differences in maternal age, race, education, welfare status, marital status, gravity, parity, ectopic pregnancies, stillbirths, delivery type, alcohol, marijuana or cigarette use (all p’s < 0.2). Women who did not return were more likely to be cocaine users (45% vs. 28%; p=0.004) and unemployed (60% vs. 47%; p= 0.02) compared to women who returned for follow-up. Infants of women who did not return were more likely to have lower birth weight (3017 g ± 512 vs. 3241 g ± 516; p 0.001) and birth length (49.5 cm ± 2.5 vs. 50.8 cm ± 2.5).
Follow-up
We invited women and their infants for follow-up at 6, 12 and 24 months of age for neurological, developmental and behavioral assessments. Of the 380 infants, 306 (80%) children returned for follow-up at least once; 286 (75%) for their 6-month assessment, 277 (73%) for their 12-month assessment and 251 (66%) for their 24-month assessment. Two hundred and twenty six children (60% of the total cohort) were seen at all 3 time points.
Measures Maternal: Maternal information including demographic, obstetrical and drug history data was be collected at the time of enrollment using a standardized questionnaire that has been successfully used in epidemiological studies [50]. These schedules ask women about obstetrical and medical histories, illnesses, smoking, alcohol use and drug use during pregnancy. The interview provides quantitative measures of alcohol and smoking, such as number of cigarettes smoked per day, number of drinks a day, type of alcoholic beverage and quantity ingested during the pregnancy. Also, binge drinking, alcohol use in the month prior knowing about pregnancy. Day by day, recall for an average week during the month before pregnancy and during mid and latter pregnancy was averaged to obtain a mean level of consumption in ounces of absolute alcohol per day (AA/day). The interviewer was trained in the administration of interview.
Caregivers were administered an IQ test that relied upon the Vocabulary and Matrix Reasoning subtests of the Wechsler Abbreviated Scale of Intelligence (WASI) [51].
At the 6-month visit, caregivers were administered a lifestyle questionnaire, the Center for Epidemiological Study-Depression Scale (CES-D) [52] and the WASI test. Because of concerns for the safety of our staff, we did not conduct home visits; instead, at 12 and 24 months we administered a version of the HOME scale [53] modified for use in the office following recommendations by Jacobson et al. [54].
Biological markers: At the time of enrollment, we collected maternal hair samples (n=380) and used segmental hair analyses (proximal 3.9 cm of hair) for radioimmunoassay for metabolites of drugs used during the 3rd trimester (RIAH™), as this could be measured in all women. All tests positive for cocaine in urine and meconium were also positive for cocaine on hair testing. Of women who denied using cocaine during pregnancy, 28% had a positive biomarker for cocaine; while of those testing positive for cocaine only 18% reported using cocaine or crack during pregnancy. Quantitative assays measured metabolites of cocaine, opiates, phenylcyclidine (PCP), amphetamines, and qualitative assays measured marijuana exposure. Hair samples were tested at Psychemedic Corp; Culver City, CA. We collected meconium in a subset of children for whom meconium specimens could be collected within the first 24 hours (N=109) and urine in a subset of mothers (N=172). Meconium and urine specimens were tested at MecStat Laboratories, Desplaines, IL.
Child measures: At 6 and 12 months of age, children were administered the full Bayley Scales of Infant Development II (BSIDII) [55] (including the Infant Behavior Rating Scale, BRS) and the Fagan Scale of Infant Intelligence [56] by staff trained by a clinical psychologist (GW). The 6 month data has been previously reported [48]. It is included this report for the sake of completeness of the developmental growth curve analyses. Inter-rater reliability was achieved between examiners (ICC > 0.95). At 24 months, we administered the mental subscale of the Bayley Scales of Infant Development II and the Peabody Motor Scale of Development [57] and obtained blood for hemoglobin and lead level. Neurological function was assessed at all age intervals with the Neurological Examination of Children [58] by a single child neurologist (CAC) who was blind to exposure status.
Exposure ascertainment: Because of the exponential range of cocaine exposure measures in hair (5-3764 ng/10 mg hair; mean 31 ng/10 mg), we used the log transformed (base 10) amount of cocaine metabolite quantified in maternal hair samples in statistical analyses to assess associations between prenatal cocaine exposure and infant outcomes. We grouped children into a cocaine-unexposed (CU) group (n = 206) if cocaine metabolites in maternal hair were below the threshold of detection (< 5 ng/10 mg), a low cocaine-exposure (CE1) group if metabolites were < 1.5 log ng/10 mg of hair and a high cocaine-exposure (CE2) group if metabolites were detected >1.5 log ng/10 mg. The median log of cocaine exposure in group CE1 was 1 and the median for CE2 was 2. For dichotomous (yes/no) exposure data, we classified infants as exposed if any biological sample tested positive for cocaine at delivery (either in urine from mother or in infant meconium).
We used alcohol equivalents (e.g. 12 oz beer, 4-5 oz of wine, wine coolers, 1 shot hard liquor) to estimate maternal alcohol intake based on the recall of number of drinks for an average week during the month prior to pregnancy and during pregnancy (early and late) to obtain a mean level of daily consumption (drinks per day). We also asked about binge drinking (5 drinks or more on one occasion). Number of average drinks per day during pregnancy was the variable most strongly associated with the outcome and was used in all analyses.
Cigarette exposure was ascertained by biomarkers (cotinine) in meconium or maternal urine in 220 subjects (72%). Based on selfreport and biomarkers, exposure was defined as light (< 5 cigarettes/day or < 50th percentile on biomarker) and heavy (> 5 cigarettes/day and > 50th percentile on biomarker). When discrepancies were noted between self-report and level of exposure found on toxicology, analyses were based on toxicology results, given maternal underreporting of drug and cigarette use during pregnancy [40,59]. Marijuana toxicology measures were qualitative. Heavier marijuana users were defined as those who tested positive on biomarkers or who reported using marijuana more than twice a week.
Statistics
Single time point analyses: For categorical variables we used either the χ2 test or Fisher’s exact test. For dichotomous neurological outcome, we used odds ratio to assess the strength of association. We used children without cocaine exposure as the base rate against which varying levels of cocaine exposure were tested. Contrasts between cocaine-unexposed children (CU; N=115) and children exposed to low (CE1; N=105) and high (CE2; 86) cocaine levels were determined by one-way analysis of variance for continuous variables and either the χ2 test or Fisher’s exact test for categorical variables. Stepwise multiple regressions were used to evaluate associations with cocaine exposure after adjusting for potential confounders. We excluded 10 opiate exposed children from all multivariate analyses. Covariates were selected based on empirical and theoretical criteria. Using empirical criteria, from the pool of potential covariates, we retained variables from Table 1 in models when they differed significantly among exposure groups and were associated with a given outcome at P ≤ 0.2. We excluded covariates if they correlated (r > 0.70) with other covariates as to preclude examination. For theoretical reasons we used, birth weight, prematurity, small for gestational age (SGA), small head size (< 10 percentile), and alcohol, tobacco, and marijuana exposure as well as the level of lead and hemoglobin in blood at the age of 2 years, as covariates in least square models. Maternal and environmental variables included education level, IQ, maternal depressive symptoms and HOME scale (12 and 24 month). To assess mediation, all analyses were run with and without the inclusion of select perinatal variables (i.e. birth weight, microcephaly, and gestational age) to determine whether their inclusion modified associations between prenatal cocaine exposure and neurodevelopmental outcome. Because of the high levels of shared variance between different covariates and outcomes, we removed covariates from a model if they failed to add significant variance to the model. The remaining covariates were included with the exposure effect of interest in linear regression models. Interaction terms were tested and not found to be significant and were thus omitted from final models. We entered level of cocaine exposure into the model at the final step to assess the impact of cocaine on overall mental, motor, novelty preference and behavioral scores while controlling for the other covariates.
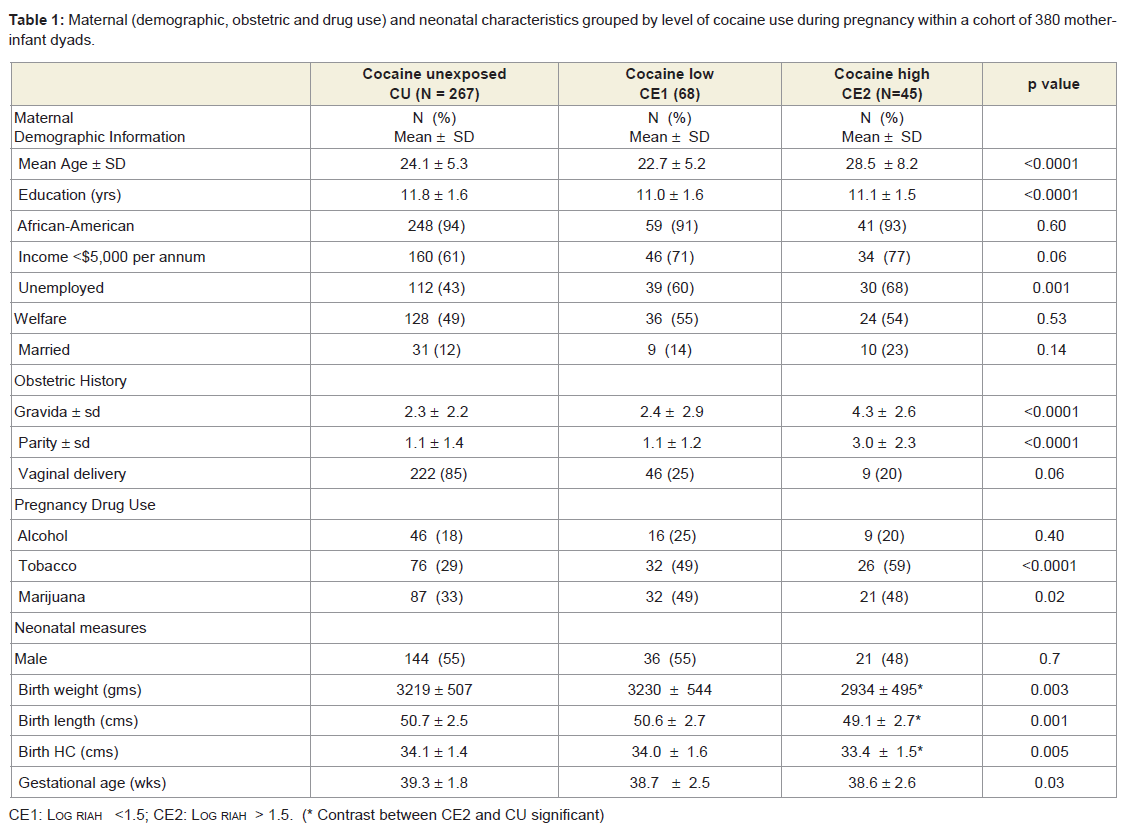
Table 1: Maternal (demographic, obstetric and drug use) and neonatal characteristics grouped by level of cocaine use during pregnancy within a cohort of 380 mother-infant dyads.
Longitudinal analyses: To assess the association between cocaine exposure with infant’s overall performance at 6, 12 and 24 months simultaneously, mixed-effects linear modeling was conducted separately for the mental developmental index (MDI) and for the behavior rating scale (BRS). Similar analyses at 6 and 12 months considered novelty preference (Fagan test of Infant Intelligence) and psychomotor development index (PDI) using SAS PROC MIXED (version 8.2; SAS Institute, Cary, NC). To allow for comparison of the different motor tests administered across ages, we transformed test scores to their respective z-scores.
The mixed-effects linear model is a generalization of the standard linear model, with the generalization being that the data can be correlated [62]. It is commonly used to analyze longitudinal data because it allows explicit modeling and analysis of between and within-individual variation and has the capacity to handle unbalanced data. The mixed-effects model is often referred to as the two-stage random-effects model; at stage 1 the population parameters and within-individual variation are introduced, and at stage 2 the between-individual variation is introduced. We first constructed a baseline model for all the infants that included an intercept term representing overall performance at 6, 12 and 24 months and a linear term representing change with age. We used the baseline model to assess the association of level of cocaine exposure with overall performance. We also examined the associations of level of cocaine and the change with age by assessing the interaction term between cocaine exposure and age.
In choosing the correlation structure, we considered unstructured, compound symmetry and first-order autoregressive structures. Analyses of 6, 12 and 24-month data showed little difference in outcome using any of these structures. We chose to analyze our data as unstructured, given the disparity between the variances of the outcome variables at different time points. Insofar as each child contributes to her own developmental curve, the randomeffects model we have chosen for this study is well suited to dampen any biases borne from differential rate of attrition based on level of cocaine exposure.
Covariate analyses: We selected covariates based on empirical and theoretical criteria. We included the following variables in initial mixed-effect model: HOME Scale, maternal depression scores, maternal IQ, maternal age < 30, welfare status, prematurity, birth weight, birth head circumference < 10th percentile, infant’s gender, tobacco, alcohol, and marijuana exposure. To assess whether hypertonia, birth weight or microcephaly (at age 6 months) mediated cocaine effects on later developmental outcome, we assessed the final model with and without these variables. We ran final models with and without birth weight, hypertonia or head size in order to determine whether these factors independently mediated any cocaine effects on neurodevelopmental outcome. The factors were chosen because of the known association between motor impairments [63], intrauterine growth retardation [64] and microcephaly [64,65] with adverse neurodevelopment. We considered hypertonia, birth weight or microcephaly to mediate cocaine effects on development (i.e. effect modification) if their inclusion in analyses lead to a significant change in the association between prenatal cocaine exposure and developmental outcome.
Results
Maternal demographic and perinatal information for the initial cohort (N= 380) is depicted in Table 1, revealing a homogenous population of women, mostly African-American (90%) and of low socioeconomic status. Cocaine-using mothers were significantly more likely to be unemployed, and less educated and to have had a higher rate of pregnancies (Table 1). Women with heavy cocaine use tended to be older than light or non-cocaine users. Comparisons between the cocaine-unexposed (CU) and the two cocaine-exposure groups (CE1 and CE2) showed higher rates of exposure to tobacco and marijuana (Table 1) in the exposed infants. Rates of alcohol use were similar between unexposed (CU) children and those in either exposure group (CE1 and CE2).Comparisons of cocaine-unexposed (CU) children with cocaineexposure level (CE1 and CE2) children showed no significant differences in age, gender, z-scores of weight, height and head circumference at the time of the 6, 12 or 24-month visit (data not shown). At birth, cocaine-exposed children had lower weight, length and head circumference when compared to unexposed infants [48]. Birth weight differences were accounted primarily by gestational age and not by cocaine exposure (data not shown).
Adjusted single time point analyses
Mental Development Index (MDI): Prenatal cocaine exposure was unrelated to 6-month MDI scores with or without adjustment for significant factors (maternal IQ, HOME scale, and infant gender; F= 3.556; p= 0.008; R2 = 0.051). In contrast, prenatal cocaine exposure was related to 12-month MDI scores, but not after adjusting for significant factors (maternal IQ, gestational age, microcephaly and infant gender; F= 5.383; p= 0.000; R2 = 0.081) (Table 2) [48]. Prenatal cocaine exposure was unrelated to 24-month MDI with or without adjustment for significant factors (maternal IQ, HOME scale, welfare status, microcephaly, sex and exposure to alcohol and tobacco; F= 4.619; p= 0.000; R2 = 0.137) (Table 2).
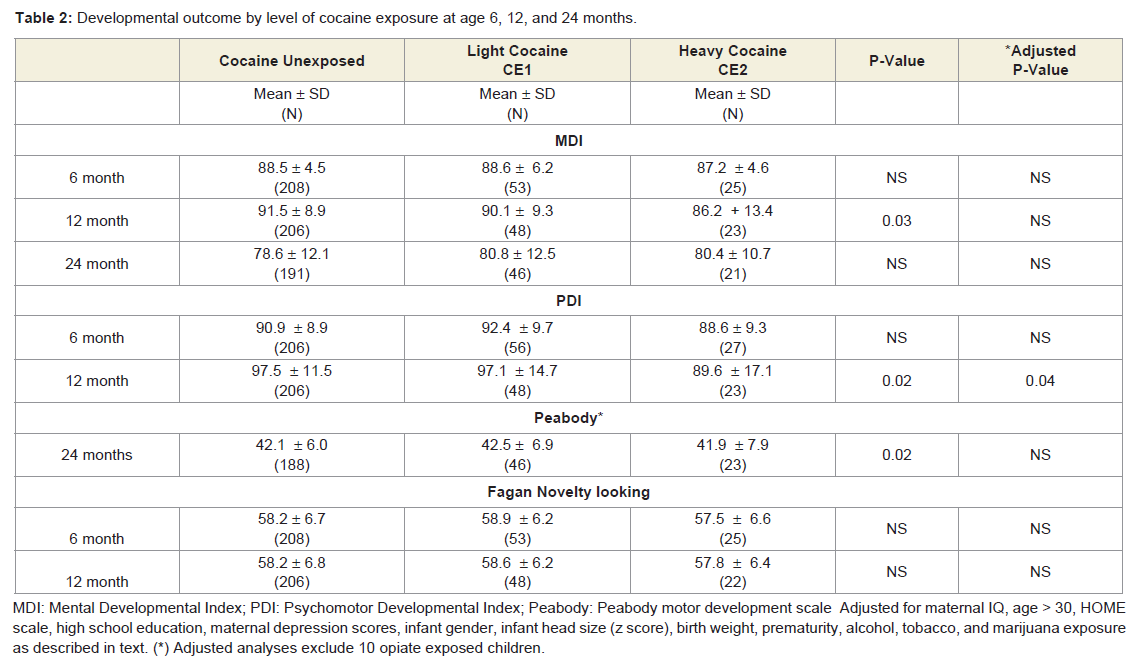
Table 2: Developmental outcome by level of cocaine exposure at age 6, 12, and 24 months.
Fagan’s Novelty preference: At age 6 months, novelty preference was unrelated to exposure with or without adjustment for maternal IQ, Apgar 5 minutes, marijuana exposure and infant head size z-score (F= 3.245; p= 0.0007; R2 = 0.061) (P = 0.84) (Table 2) [48]. And at age 12 months, novelty preference was unrelated to prenatal cocaine exposure with or without adjustment for gestational age, maternal depression score, birth weight and income < $5,000 (F= 2.777; p = 0.018; R2 = 0.051).
Behavioral outcome: At the age of 6 months prenatal cocaine exposure was marginally associated with differences in the Bayley Total Behavior Rating Scale with and without adjustment for birth weight (p = 0.06; .-3.8 points; ΔR square 0.014) (Table 3) [48]. Prenatal cocaine exposure was also significantly associated with Orientation/Engagement scores with (p = 0.04) and without adjustment (p= 0.01) for maternal IQ and maternal age > age 30; (F= 4.19; p =0.006; R2 =0.045; -3.8 points; ΔR square =0.019) (Table 3)). At 12 and 24 months of age, prenatal cocaine exposure was unrelated to scores on the Behavior Rating Scale, or on either the Orientation/Engagement or the Emotional Regulation scale with adjustment of significant factors. The 12-month unadjusted Orientation/Engagement scale was related to cocaine exposure (p=0.02) but not after adjusting for alcohol and tobacco exposure (F= 5.251; p = 0.006; R2 = 0.038) (Table 3).
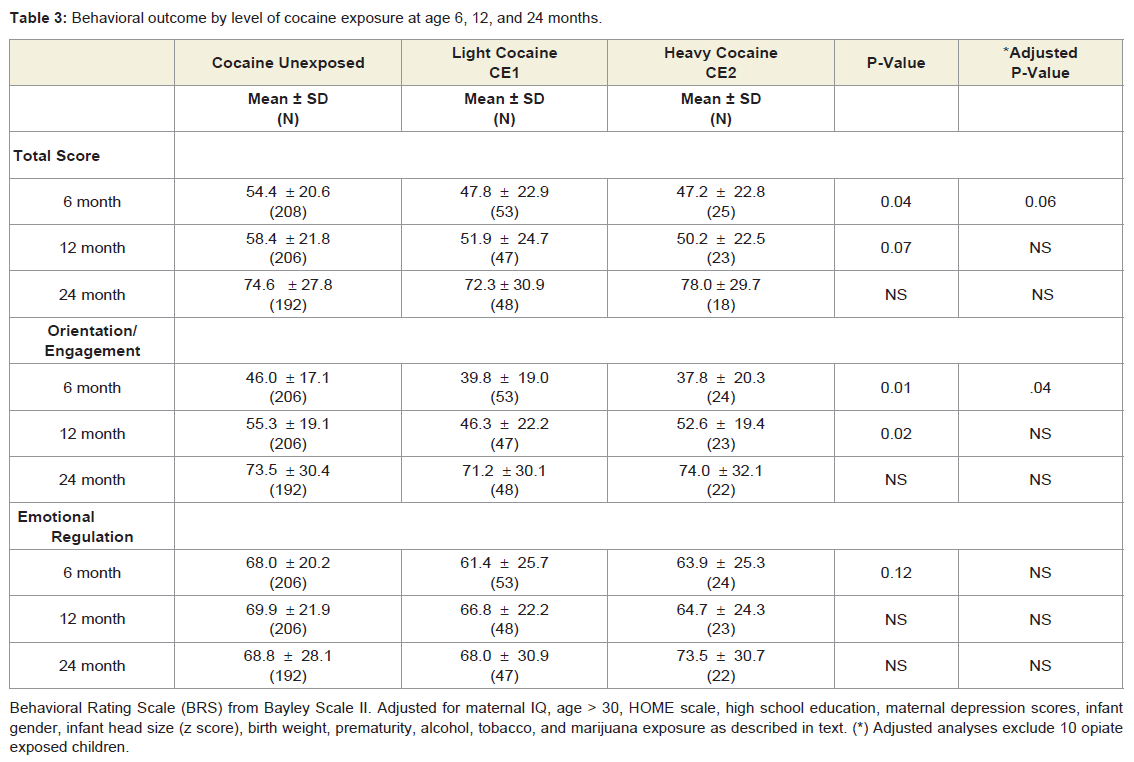
Table 3: Behavioral outcome by level of cocaine exposure at age 6, 12, and 24 months.
Bayley scales of infant development-II mental (MDI) and motor index (PDI): Table 4 shows the parameter estimates for the age and level of cocaine exposure. The mental index decreased across age for all children. The average mental index in adjusted analyses (sex and maternal IQ) was significantly lower (-2.2 points) for infants with the higher level of cocaine exposure compared to unexposed infants (p = 0.04). A significant interaction was observed between the age and level of cocaine exposure (β = 0.12, p = 0.013), indicating that the average curves for cocaine-exposed and unexposed children were different across age. Cocaine exposure was associated with a more negative slope than the slope among unexposed. Microcephaly at the age of 6 months, when introduced into the final equation, was marginally related to MDI (-2.9 points; p= 0.056) and altered the association between cocaine exposure and mental development (cocaine exposure p= 0.17). Hypertonia at 6 months of age was not associated with longitudinal developmental scores (p > 0.1).
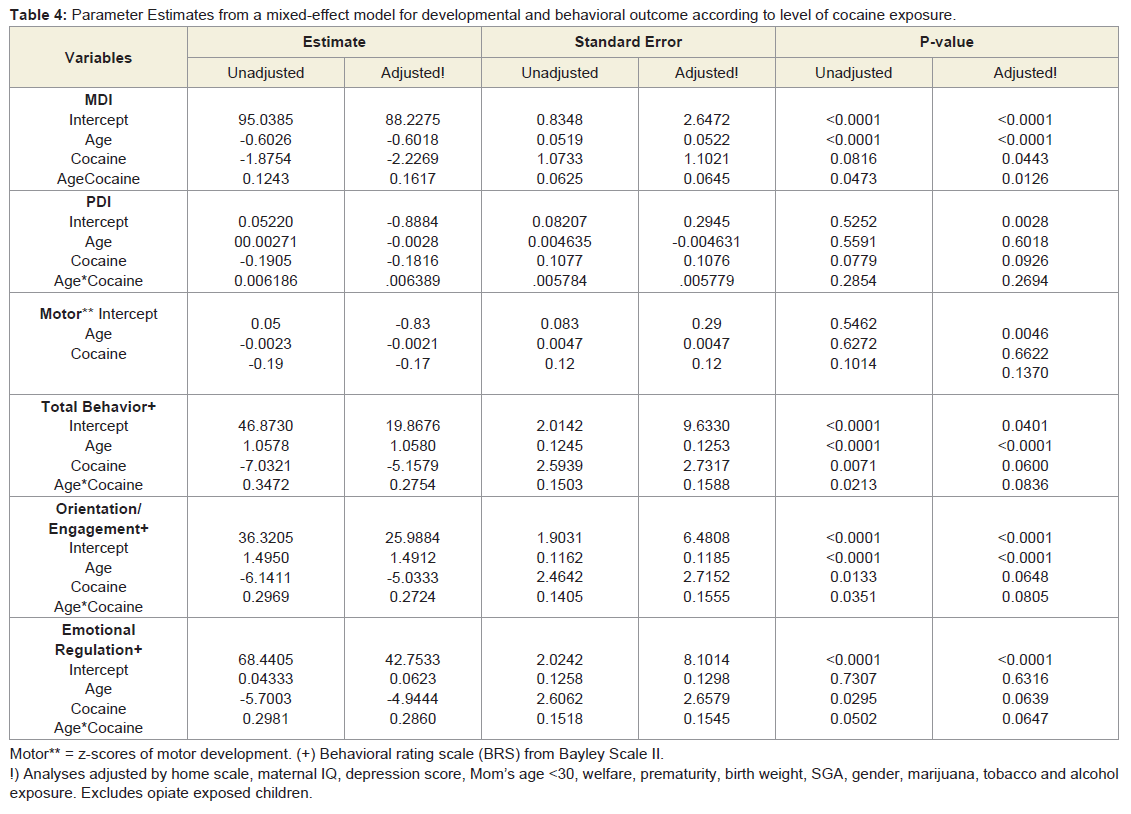
Table 4: Parameter Estimates from a mixed-effect model for developmental and behavioral outcome according to level of cocaine exposure.
Behavior rating scale: Cocaine exposure was marginally associated (p = 0.06) with all of the behavioral outcomes in analyses that adjusted for the following; Total Behavior: birth weight, maternal education and alcohol exposure; Orientation/Engagement: maternal IQ and alcohol exposure; Emotional Regulation: maternal education, birth weight, tobacco exposure and small for gestational age. Age was significantly associated (p < 0.0001) with Total Behavior scores and Orientation/Engagement; the positive slope of age for these outcomes indicates improvement with age.
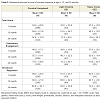
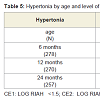
Neurological outcome: Table 5 shows the rates of hypertonia (global and diparesis) identified at the ages 6, 12 and 24 months. Rates were highest at the 6-month visit across all levels of exposure. In this cohort, we reported previously a significant association between levels of cocaine exposure and rates of hypertonia at 6 months of age that remained after controlling for confounding variables including other perinatal exposures (see [48]). Rates of hypertonia noted at 6 months dropped dramatically by the 12-month visit. Because the survival analysis focused on the evolution of hypertonia noted at age 6 months, findings of hypertonia identified de novo in 4 unexposed children at age 24 months were not included in any of the survival analyses. Figure 1 shows the survival curve for resolution of hypertonia (Kaplan Meier proportional hazards) showing a more rapid rate of resolution among cocaine-exposed children. In Cox regression models controlling for prematurity and gender, each log of cocaine exposure was associated with a 1.5 fold increased odds (Hazard Ratio, HR) of improvement (p = 0.01). In similar analyses that assessed for dichotomous heavy and light cocaine exposure, hypertonia improved 2.2 times faster (HR) among those with heavy exposure (95% confidence interval 1.1 - 4.4; p = 0.02), and 1.5 faster (HR) among those with light exposure (95% confidence interval 0.8 - 2.7; p = 0.18).
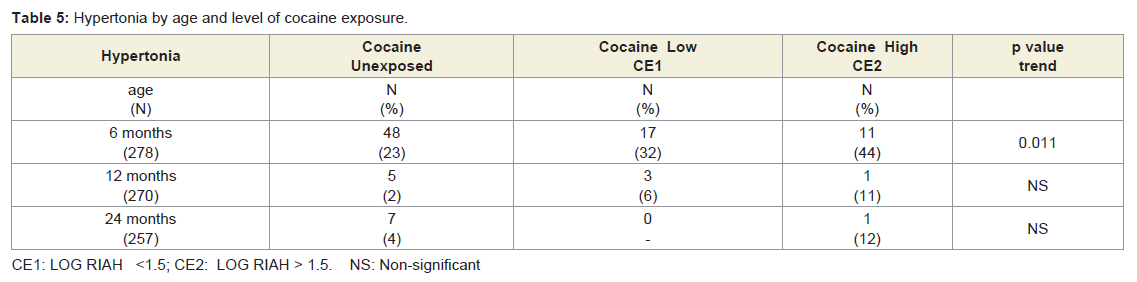
Table 5: Hypertonia by age and level of cocaine exposure.
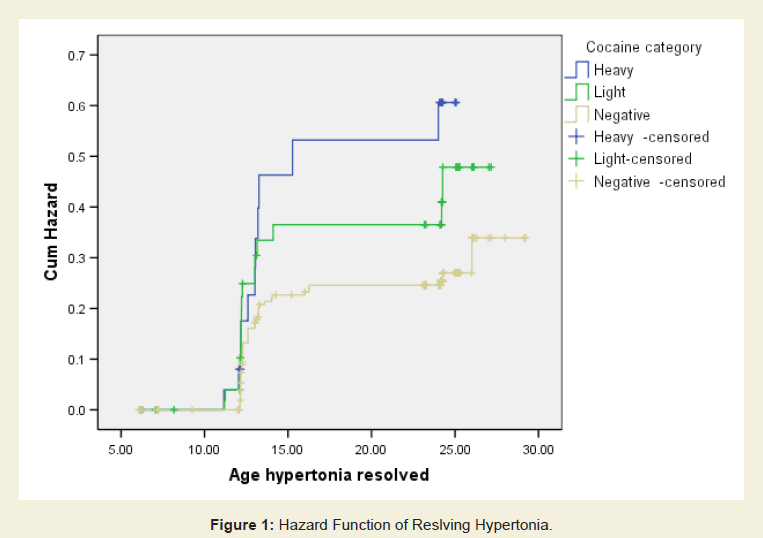
Figure 1: Hazard function of resolving hypertonia.
Discussion
We describe the developmental, behavioral, and neurological outcomes associated with levels of prenatal cocaine exposure in an urban cohort followed over the first 2 years of life in whom prenatal drug exposures was carefully ascertained with a variety of biomarkers. Although cocaine exposure ascertainment based on hair reflects the last trimester of pregnancy, it is assumed that exposure from cocaine occurred throughout pregnancy as few women would be expected to begin using cocaine de novo after becoming pregnant.Development
The developmental findings that emerge from this study are: 1) a significantly negative slope in mental development growth trajectory, but not in motor development, with advancing age; 2) significantly different slopes in mental development trajectories with cocaineexposed children showing significantly greater negative slopes than cocaine-unexposed children and 3) a negative slope in mental performance associated with prenatal cocaine exposure that was mediated by microcephaly. A decline with age in mental development on the Bayley scales (i.e. negative slope in mental trajectory) is well documented, especially by the 24 months assessment [66], and likely reflects the increasing dependence of the test on language skills by the age of 24 months. Language skills in turn are sensitive to environmental factors (e.g. poverty, maternal intelligence or education). As noted, prenatal cocaine exposure affects language [5-7]. As to why effects were noted on mental development trajectories but not on single time point analyses is not clear, but may reflect the robustness of the developmental growth curve, in which each subject contributes several time points to the model reflecting changes over time, achieving, thus, greater statistical power than possible with a single observation.
Our findings of delays in mental development linked to prenatal cocaine exposure are congruent with findings of other single time point analyses [1-3]. Several developmental trajectory studies have been undertaken [4,11,67] but only 2 in age range depicted here [11,67]. Mayes et al. noted mental delays with a decline in trajectory through 24 months, though effects were not significant in adjusted analyses [67]. Effects were mediated by birth weight and gestational age, but the study did not control for head size. The authors hypothesized that nutritional factors, perhaps related to cocaine’s known vasoconstrictive properties, mediated effects. Similarly, Miller-Loncar et al. also found effects related to cocaine exposure on motor development that disappeared after controlling for birth weight and environmental factors [11]. The current analyses controlled for biological and environmental factors, as well as for other perinatal exposure variables (alcohol, tobacco and marijuana). Of the more than 20 variables assessed in the initial model, other than cocaine, only maternal IQ and sex were associated with mental development. In this study, unlike findings reported by Mayes et al. [67], anthromorphic birth measures (i.e. microcephaly at birth, SGA, birth weight and prematurity) did not mediate effects on developmental outcome.
Cocaine is amply reported to impair fetal growth [40,68,69]. Yet the effects of cocaine on neonatal growth, unlike findings with alcohol [70] (especially fetal alcohol syndrome) [71] tends to be transient, with many studies noting postnatal catch-up growth [72-74], at least in the age range targeted for study herein (although not at later ages) [74]. Such was the case in this cohort, in which the somatic and head growth impairments present at birth linked to cocaine exposure resolved by age 6 months: we found no differences in weight or height z-scores based on the level of cocaine exposure at any age, after the newborn period [48].
One could postulate, as did Mayes et al. [67], that differences in mental development could arise from environmental differences linked to cocaine use, such as the quality of caregiver interaction [75,76], number of caregivers or ongoing cocaine use. Although we controlled for environmental variables, including maternal IQ, depression scores, HOME Scale, education and welfare status, these measures undoubtedly adjust only imperfectly for the adverse impact of a non-supportive environment. However, our findings of mediation by infant microcephaly would imply that biological, not environmental factors, account for differences in mental performance associated with prenatal cocaine exposure. Similar mediation of developmental effects by head size has been previously reported [77].
It is of interest that microcephaly at the age of 6 months, but not at birth, mediated cocaine effects. This finding would support the notion that cocaine effects on development are not nutritional in nature (as newborn size reflects maternal and placental nutrition [78]), but instead indicative of a sustained embryopathic effect on brain development. Experimentally, prenatal cocaine exposure has been shown to inhibit proliferaton of neural progenitors [79] and result in microcerebral dysgenesis [80-82] with thinner cortices and fewer neurons [81] noted in animals that also exhibit smaller brain volumes [80]. Teratogenic effects of cocaine on brain development have implicated GABA systems, with cocaine exposure in early neurogenesis found to impair radial migration of GABA and glutamatergic neurons [82]. This teratogenic effect of cocaine on brain development could be the underlying mechanism by which small head size mediates cocaine effects on mental development. Verifying this possibility in humans is stymied by the lack of neuropathological data among the population of interest. In a single case report, microcephaly was noted in an infant with documented prenatal cocaine exposure whose brain showed migrational deficits that mirrored experimental findings [83].
Many perinatal studies have not controlled for head circumference, a surrogate marker of brain size [67]. Others have controlled for head size using z-scores [15]. We have chosen instead to control for head size using microcephaly (i.e. children’s head size that fall at the lower end of the Gaussian distribution). Although head size that falls at the lower end of the curve may simply be constitutionally small (i.e. normal brains of smaller size), such head sizes are much more likely in children with pathologically small heads, and thus is more strongly linked to lower cognition [64,65,84]. In contrast, cognitive abilities among children with head sizes in the middle and upper parts of the Bell curve are expected to be independent of head circumference. The practice of controlling for small head size, however, may be inappropriate if this outcome, as our study suggests, lies in the causal pathway of cocaine’s detrimental effect on development.
Behavior
As regards neurobehavioral findings, we reported at the age of 6 months significant associations between prenatal level of cocaine exposure and hypertonia and behavioral outcome (Orientation/Engagement) with a trend with Total Behavior (p=0.06) in adjusted analyses [48]. Similar cocaine-related behaviors have been reported in young infants, such as excessive irritability at an age of 3 months [25]. Here we set out to determine the behavioral trajectory and evolution of these early neurological findings. Behavior trajectories in all domains were lower among cocaine-exposed children but only marginally so in adjusted analyses and tended to improve over time, especially among cocaine-exposed children, as indicated by the significant interaction between age and cocaine in unadjusted analyses.
Neurological
Neurological findings of hypertonia also improved with age. These findings are consistent with an earlier report describing transient high rates of hypertonia associated with cocaine exposure among infants at risk for HIV infection that resolved by 24 months [46]. Children in that cohort, however, exhibited higher rates of hypertonia than is described herein, and those higher rates of hypertonia lingered into the 12-month assessment, perhaps reflecting the higher level of maternal addiction encountered among HIV infected women. The transient nature of hypertonia noted is in agreement with the improvement in motor development reported over the first 18 months of life. We noted a cocaine effect on motor development at 12 months of age, and a non-significant trend (p = 0.07) on early motor trajectory (6 & 12 months) both of which are consistent with motor delays reported among cocaine-exposed infants and toddlers [8-11]. The absence of effect on motor development found in our 2 year trajectory may be a reflection of the older age of follow-up, after motor deficits resolve.
We postulated, at the onset of the study that cocaine-related hypertonia could adversely affect development. Our results do not support this perspective. Although hypertonia was negatively correlated with early psychomotor development, it had no effect on either the mental or the motor developmental trajectory (i.e. no effect on development at age 2 years). In contrast to our mental development findings, behavioral and neurological findings were prominent in early infancy and improved over time.
It is conceivable that the transient neurobehavioral manifestations observed herein are due to transient embryopathic perturbation of DA and GABA systems. Experimentally, in utero cocaine exposure reduces dopamine-dependent reflexes and motor task performance in mice [24]. Although in utero cocaine exposure affects DA systems, whether DA has a direct role in eliciting abnormal behaviors has been questioned [85]. Increasingly, cocaine exposure in utero has been noted to alter the structure and function of the postnatal cerebral cortex though effects on GABA circuits [24,82,86,87]. The findings by Lu of elevated neuronal excitability on pyramidal neurons also attributed to reduced GABAergic inhibition system may explain motor findings noted in this study (motor delay and hypertonia) [86].
Postnatally, over time, such perturbations would tend toward homeostasis with concomitant resolution of earlier impairments. Such a model would explain why hypertonia among cocaine exposed children, especially those more heavily exposed, recovered at a faster rate and with a lower base rate at age 24 months than in unexposed children. Among the latter, hypertonia presumably would have a different etiology (e.g. vascular or inflammatory) that might not be self-correcting. One could thus envision two different mechanisms operant - one related to perturbations in neurotransmitter function causing excitability and motor impairments transiently, and the other related to migrational impairments affecting head growth and cognition that would be long lasting.
Insofar as study participants were able to keep custody of their children and maintain follow-up appointments, they likely differ from mothers at the higher end of the cocaine-addiction spectrum. On the contrary, cocaine-exposed women with offspring showing greater newborn growth impairments were more likely to be lost to follow-up. Differential loss to follow-up may have biased our study towards the null, as possibly more heavily cocaine-exposed women were not included in the longitudinal analyses. Such type II errors tend to decrease the strength of associations and thus, underestimate effects. Our study differs from others in several respects. A key factor is the use of hair to quantify cocaine exposure. Although hair measures have been criticized because of possibility of external contamination by ambient cocaine (which is avoided by strict hair washing protocols) we have found hair, in this [48] and in earlier studies [40,88], to be an excellent method by which to quantify exposure and assess dose response effects. As a continuous measure, it also provides greater power than dichotomous methods of exposure ascertainment, allowing us to determine associations that would not otherwise be apparent with the present modest sample size. Further, our use of a structured neurological instrument allows us to describe the evolution of neurological findings as pertains to specific domains (tone, reflexes etc.) and to correlate findings with developmental outcome. We controlled for lead and hemoglobin at age 2 years because both are known to affect infant cognition [89].
One limitation in this and in other clinical studies, is that despite our best efforts at quantifying and controlling for other substances, cocaine effects may be confounded by polydrug exposure. Alcohol is a case in point where, because of stigma associated with alcohol intake during pregnancy, the veracity of maternal report may be affected. Despite our efforts to carefully assess alcohol intake during and prior to known pregnancy, because this measure relies exclusively on interview and has no reliable biochemical marker we may have underestimated the level of alcohol exposure. Such polydrug exposure could exert synergistic effects on head growth; conceivably prenatal cocaine exposure may be simply one of many adverse perinatal factors that may impact brain development in high risk cohorts. Insofar as our examiner ratings of behavior were restricted to testing in a single environment (study visit), the generalizability of behavioral findings is also a potential study limitation [90]. Other limitations may include enrollment of infants at birth and the generalizability of findings of a predominant African-American population. The young age of our cohort precludes any determinations regarding ultimate behavior, intellectual, and learning abilities associated with prenatal cocaine exposure. Such determinations will require long-term follow-up.
Acknowledgements
We wish to thank Liang Li and Dr. Yuqing Yang for their biostatistical assistance in analyzing the data.Funded by NIDA, R01-DA011410.References
- Alessandri SM, Bendersky M, Lewis M (1998) Cognitive functioning in 8- to 18-month-old drug-exposed infants. Dev Psychol 34: 565-573.
- Singer LT, Arendt R, Minnes S, Farkas K, Salvator A, et al. (2002) Cognitive and motor outcomes of cocaine-exposed infants. JAMA 287: 1952-1960
- Pulsifer MB, Butz AM, O’Reilly Foran M, Belcher HM (2008) Prenatal drug exposure: effects on cognitive functioning at 5 years of age. Clin Pediatr (Phila) 47: 58-65.
- Bennett DS, Bendersky M, Lewis M (2008) Children's cognitive ability from 4 to 9 years old as a function of prenatal cocaine exposure, environmental risk, and maternal verbal intelligence. Dev Psychol 44: 919-928.
- Singer LT, Arendt R, Minnes S, Salvator A, Siegel AC, et al. (2001) Developing language skills of cocaine-exposed infants. Pediatrics 107: 1057-1064.
- Morrow CE, Vogel AL, Anthony JC, Ofir AY, Dausa AT et al. (2004) Expressive and receptive language functioning in preschool children with prenatal cocaine exposure. J Pediatr Psychol 29: 543-554.
- Lewis BA, Singer LT, Short EJ, Minnes S, Arendt R, et al. (2004) Four-year language outcomes of children exposed to cocaine in utero. Neurotoxicol Teratol 26: 617-627.
- Richardson GA, Goldschmidt L, Willford J (2008) The effects of prenatal cocaine use on infant development. Neurotoxicol Teratol 30: 96-106.
- Arendt R, Angelopoulos J, Salvator A, Singer L (1999) Motor development of cocaine-exposed children at age two years. Pediatrics 103: 86-92.
- Fetters L, Tronick EZ (1996) Neuromotor development of cocaine-exposed and control infants from birth through 15 months: poor and poorer performance. Pediatrics 98: 938-943.
- Miller-Loncar C, Lester BM, Seifer R, Lagasse LL, Bauer CR, et al. (2005) Predictors of motor development in children prenatally exposed to cocaine. Neurotoxicol Teratol 27: 213-220.
- Frank DA, Augustyn M, Knight WG, Pell T, Zuckerman B (2001) Growth, development, and behavior in early childhood following prenatal cocaine exposure: a systematic review. JAMA 285: 1613-1625.
- Ackerman JP, Riggins T, Black MM (2010) A review of the effects of prenatal cocaine exposure among school-aged children. Pediatrics 125: 554-565.
- Hurt H, Betancourt LM, Malmud EK, Shera DM, Giannetta JM, et al. (2009) Children with and without gestational cocaine exposure: a neurocognitive systems analysis. Neurotoxicol Teratol 31: 334-341.
- Wasserman G, Kline JK, Bateman DA, Chiriboga CA, Lumey LH, et al. (1998) Prenatal cocaine exposure and school-age IQ. Drug Alcohol Depend 50: 203-210.
- Richardson GA, Conroy ML, Day NL (1996) Prenatal cocaine exposure: effects on the development of school-age children. Neurotoxicol Teratol 18: 627-634.
- Minnes S, Singer LT, Min MO, Lang AM, Ben-Harush A, et al. (2014) Comparison of 12-year-old children with prenatal exposure to cocaine and non-exposed controls on caregiver ratings of executive function. J Youth Adolesc 43: 53-69.
- Eyler FD, Warner TD, Behnke M, Hou W, Wobie K, et al. (2009) Executive functioning at ages 5 and 7 years in children with prenatal cocaine exposure. Dev Neurosci 31: 121-136.
- Singer LT, Nelson S, Short E, Min MO, Lewis B, et al. (2008) Prenatal cocaine exposure: drug and environmental effects at 9 years. J Pediatr 153: 105-111.
- Bandstra ES, Morrow CE, Accornero VH, Mansoor E, Xue L, et al. (2011) Estimated effects of in utero cocaine exposure on language development through early adolescence. Neurotoxicol Teratol 33: 25-35.
- Beeghly M, Martin B, Rose-Jacobs R, Cabral H, Heeren T, et al. (2006) Prenatal cocaine exposure and children's language functioning at 6 and 9.5 years: moderating effects of child age, birthweight, and gender. J Pediatr Psychol 31: 98-115.
- Lewis BA, Minnes S, Short EJ, Min MO, Wu M, et al. (2013) Language outcomes at 12 years for children exposed prenatally to cocaine. J Speech Lang Hear Res 56: 1662-1676.
- Giustino A, Cuomo V, Marsden CA (1998) Maternal cocaine exposure alters mesolimbic dopaminergic function in rat offspring. Eur J Pharmacol 345: 175-180.
- Wang W, Nitulescu I, Lewis JS, Lemos JC, Bamford IJ et al. (2013) Overinhibition of corticostriatal activity following prenatal cocaine exposure. Ann Neurol 73: 355-369.
- Mayes LC, Bornstein MH, Chawarska MA, Granger RH (1995) Information processing and developmental assessments in 3-month old infants exposed to cocaine. Pediatrics 95: 539-545.
- Karmel BZ, Gardner JM (1996) Prenatal cocaine exposure effects on arousal-modulated attention during the neonatal period. Dev Psychobiol 29: 463-480.
- Chiriboga CA, Starr D, Kuhn L, Wasserman G (2009) Prenatal cocaine exposure and prolonged focused attention: poor infant information processing or precocious development of attentional systems? Dev Neurosci 31: 149-158.
- Lester BM, Bagner DM, Liu J, LaGasse LL, Seifer R, et al. (2009) Infant neurobehavioral dysregulation: behavior problems in children with prenatal substance exposure. Pediatrics 124: 1355-1362.
- Bendersky M, Lewis M (1998) Prenatal cocaine exposure and impulse control at two years. Ann N Y Acad Sci 846: 365-367.
- Bridgett DJ, Mayes LC (2011) Development of inhibitory control among prenatally cocaine exposed and non-cocaine exposed youths from late childhood to early adolescence: The effects of sex and risk and subsequent aggressive behavior. Neurotoxicol Teratol 33: 47-60.
- Bendersky M, Gambini G, Lastella A, Bennett DS, Lewis M (2003) Inhibitory motor control at five years as a function of prenatal cocaine exposure. J Dev Behav Pediatr 24: 345-351.
- Ackerman JP, Llorente AM, Black MM, Ackerman CS, Mayes LA, et al. (2008) The effect of prenatal drug exposure and caregiving context on children's performance on a task of sustained visual attention. J Dev Behav Pediatr 29: 467-474.
- Delaney-Black V, Covington C, Templin T, Ager J, Nordstrom-Klee B, et al. (2000) Teacher-assessed behavior of children prenatally exposed to cocaine. Pediatrics 106: 782-791.
- Bada HS, Bann CM, Bauer CR, Shankaran S, Lester B, et al. (2011) Preadolescent behavior problems after prenatal cocaine exposure: relationship between teacher and caretaker ratings (Maternal Lifestyle Study). Neurotoxicol Teratol 33: 78-87.
- Bennett D, Bendersky M, Lewis M (2007) Preadolescent health risk behavior as a function of prenatal cocaine exposure and gender. J Dev Behav Pediatr 28: 467-472.
- Min MO, Minnes S, Lang A, Weishampel P, Short EJ, et al. (2014) Externalizing behavior and substance use related problems at 15 years in prenatally cocaine exposed adolescents. J Adolesc 37: 269-279.
- Accornero VH, Anthony JC, Morrow CE, Xue L, Mansoor E, et al. (2011) Estimated effect of prenatal cocaine exposure on examiner-rated behavior at age 7 years. Neurotoxicol Teratol 33: 370-378.
- Li Z, Santhanam P, Coles CD, Lynch ME, Hamann S, et al. (2011) Increased "default mode" activity in adolescents prenatally exposed to cocaine. Hum Brain Mapp 32: 759-770.
- Betancourt LM, Yang W, Brodsky NL, Gallagher PR, Malmud EK, et al. (2011) Adolescents with and without gestational cocaine exposure: Longitudinal analysis of inhibitory control, memory and receptive language. Neurotoxicol Teratol 33: 36-46.
- Chiriboga CA, Brust JC, Bateman D, Hauser WA (1999) Dose-response effect of fetal cocaine exposure and newborn neurologic function. Pediatrics 103: 79-85.
- Delaney-Black V, Covington C, Ostrea E Jr, Romero A, Baker D (1996) Prenatal cocaine and neonatal outcome: evaluation of dose-response relationship. Pediatrics 98: 735-740.
- Morrow CE, Bandstra ES, Anthony JC, Ofir AY, Xue L, et al. (2001) Influence of prenatal cocaine exposure on full-term infant neurobehavioral functioning. Neurotoxicol Teratol 23: 533-544.
- Tronick EZ, Frank DA, Cabral H, Mirochnick M, Zuckerman B (1996) Late dose-response effects of prenatal cocaine exposure on newborn neurobehavioral. Pediatrics 98: 76-83.
- Hurt H, Brodsky NL, Betancourt L, Braitman LE, Malmud E, et al. (1995) Cocaine-exposed children: follow-up through 30 months. J Dev Behav Pediatr 16: 29-35.
- Lewis MW, Misra S, Johnson HL, Rosen TS (2004) Neurological and developmental outcomes of prenatally cocaine-exposed offspring from 12 to 36 months. Am J Drug Alcohol Abuse 30: 299-320.
- Chiriboga CA, Vibbert M, Malouf R, Suarez MS, Abrams EJ, et al. (1995) Neurological correlates of fetal cocaine exposure: transient hypertonia of infancy and early childhood. Pediatrics 96: 1070-1077.
- Belcher HM, Shapiro BK, Leppert M, Butz AM, Sellers S, et al. (1999) Sequential neuromotor examination in children with intrauterine cocaine/polydrug exposure. Dev Med Child Neurol 41: 240-246.
- Chiriboga CA, Kuhn L, Wasserman GA (2007) Prenatal cocaine exposures and dose-related cocaine effects on infant tone and behavior. Neurotoxicol Teratol 29: 323-330.
- Lester BM, Tronick EZ, LaGasse L, Seifer R, Bauer CR, et al. (2002) The maternal lifestyle study: effects of substance exposure during pregnancy on neurodevelopmental outcome in 1-month-old infants. Pediatrics 110: 1182-1192.
- Ness RB, Grisso JA, Hirschinger N, Markovic N, Shaw LM, et al. (1999) Cocaine and tobacco use and the risk of spontaneous abortion. N Engl J Med 340: 333-339.
- Wechsler D (1999) Wechsler Abbreviated Scale of Intelligence (WASI). Psychological Corporation, San Antonio, TX.
- Radloff LS (1977) The CES-D scale: a self-report depression scale for research in the general population. Appl Psychol Meas 1: 385-401.
- Bradley RH, Caldwell BM, Rock SL, Ramey CT, Barnard KE, et al. (1989) Home environment and cognitive development in the first 3 years of life: A collaborative study involving six sites and three ethnic groups in North America. Dev Psychol 25: 217-235.
- Jacobson SW, Jacobson JL, Sokol RJ, Martier SS, Chiodo LM (1996) New evidence for neurobehavioral effects of in utero cocaine exposure. J Pediatr 129: 581-590.
- Bayley N (1993) Bayley scales of infant development (2nd ed.), Psychological Corporation, San Antonio, TX.
- Fagan JF, Singer LT (1983) Infant recognition memory as a measure of intelligence, L. P. Lipsitt (Ed.), Ablex, Norwood, NJ.
- Folio MR, Fewell RR (2000) Peabody developmental motor scales (2nd ed.), Austin, Texas.
- Chiriboga CA, Kairam R, Kline J (1993) A neurological examination for children: Reliability and utility in studies of HIV infection. Pediatr AIDS HIV Fetus Adolesc 4: 144-150.
- Kline J, Ng SK, Schittini M, Levin B, Susser M (1997) Cocaine use during pregnancy: sensitive detection by hair assay. Am J Public Health 87: 352-358.
- Kaplan EL, Meier P (1958) Non parametric estimations from incomplete observations. J Am Stat Assoc 53: 457-481.
- Cox DR, Oakes D (1984) Analyses of survival data. (Monographs on Statistics and Applied Probability), Chapman & Hall/CRC, London.
- Laird N, Ware JH (1982) Random-effects models for longitudinal data. Biometrics 38: 939-974.
- Fjørtoft T, Grunewaldt KH, Løhaugen GC, Mørkved S, Skranes J, et al. (2013) Assessment of motor behaviour in high-risk-infants at 3 months predicts motor and cognitive outcomes in 10 years old children. Early Hum Dev 89: 787-793.
- Hack M, Breslau N, Weissman B, Aram D, Klein N, et al. (1991) Effect of very low birth weight and subnormal head size on cognitive abilities at school age. N Engl J Med 325: 231-237.
- Chiriboga CA, Kuban KC, Durkin M, Hinton V, Kuhn L, et al. (2003) Factors associated with microcephaly at school age in a very-low-birthweight population. Dev Med Child Neurol 45: 796-801.
- Garcia Coll C, Buckner JC, Brooks MG, Weinreb LF, Bassuk EL (1998) The developmental status and adaptive behavior of homeless and low-income housed infants and toddlers. Am J Pub Health 88: 1371-1374.
- Mayes LC, Cicchetti D, Acharyya S, Zhang H (2003) Developmental trajectories of cocaine-and-other-drug-exposed and non-cocaine-exposed children. J Dev Behavioral Pediatr 24: 323-325.
- Zuckerman B, Frank DA, Hingson R, Amaro H, Levenson SM, et al. (1989) Effects of maternal marijuana and cocaine on fetal growth. N Eng J Med 320: 762-768.
- Shankaran S, Das A, Bauer CR, Bada HS, Lester B, et al. (2004) Association between patterns of maternal substance use and infant birth weight, length, and head circumference. Pediatrics 114: e226-e234.
- Cornelius MD, Goldschmidt L, Day NL, Larkby C (2002) Alcohol, tobacco and marijuana use among pregnant teenagers: 6-year follow-up of offspring growth effects. Neurotoxicol Teratol 24: 703-710.
- Clarren SK, Smith DW (1978) The fetal alcohol syndrome. N Engl J Med 298: 1063-1067.
- Harsham J, Keller JH, Disbrow D (1994) Growth patterns of infants exposed to cocaine and other drugs in utero. J Am Diet Assoc 94: 999-1007.
- Shankaran S, Das A, Bauer CR, Bada HS, Lester BM, et al. (2011) Prenatal cocaine exposure and small-for-gestational-age status: effects on growth at 6 years of age. Neurotoxicol Teratol 33: 575-581.
- Richardson GA, Goldschmidt L, Larkby C (2007) Effects of prenatal cocaine exposure on growth: a longitudinal analysis. Pediatrics 120: e1017-e1027.
- Burns K, Chethik L, Burns WJ, Clark R (1991) Dyadic disturbances in cocaine-abusing mothers and their infants. J Clin Psychol 47: 316-319.
- Beckwith L, Howard J, Espinosa M, Tyler R (1999) Psychopathology, mother-child interaction, and infant development: substance-abusing mothers and their offspring. Dev Psychopathol 11: 715-725.
- Azuma S, Chasnoff IJ (1993) Outcome of children prenatally exposed to cocaine and other drugs: a path analysis of three year data. Pediatrics 92: 396-402.
- Forsum E, Lof M, Olausson H, Olhager E (2006) Maternal body composition in relation to infant birth weight and subcutaneous adipose tissue. Br J Nutr 96: 408-414.
- Lee CT, Chen J, Hayashi T, Tsai SY, Sanchez JF, et al. (2008) A mechanism for the inhibition of neural progenitor cell proliferation by cocaine. PLoS Med 5: e117.
- Gressens P, Kosofsky B, Evrard P (1992) Cocaine-induced disturbances in corticogenesis in the developing murine brain. Neurosci Lett 140: 113-116.
- Ren JQ, Malanga CJ, Tabit E, Kosofsky BE (2004) Neuropathological consequences of prenatal cocaine exposure in the mouse. Int J Dev Neurosci 22: 309-320.
- Lee CT, Chen J, Worden LT, Freed WJ (2011) Cocaine causes deficits in radial migration and alters the distribution of glutamate and GABA neurons in the developing rat cerebral cortex. Synapse 65: 21-34.
- Kesrouani A, Fallet C, Vuillard E, Jacqz-Aigrain E, Sibony O, et al. (2001) Pathologic and laboratory correlation in microcephaly associated with prenatal cocaine exposure. Early Hum Dev 63: 79-81.
- Gross SJ, Kosmetatos N, Grimes CT, Williams ML (1978) Newborn head size and neurological status. Predictors of growth and development of low birth weight infants. Am J Dis Child 132: 753-756.
- Phillips PE, Johns JM, Lubin DA, Budygin EA, Gainetdinov RR, et al. (2003) Presynaptic dopaminergic function is largely unaltered in mesolimbic and mesostriatal terminals of adult rats that were prenatally exposed to cocaine. Brain Res 961: 63-72.
- Lu H, Lim B, Poo MM (2009) Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci 29: 12664-12674.
- Stanwood GD, Washington RA, Levitt P (2001) Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cereb Cortex 11: 430-440.
- Bateman DA, Chiriboga CA (2000) Dose-response effect of cocaine on newborn head circumference. Pediatrics 106: E33.
- Wasserman G, Graziano JH, Factor-Litvak P, Popovac D, Morina N, et al. (1992) Independent effects of lead exposure and iron deficiency anemia on developmental outcome at age 2 years. J Pediatr 121: 695-703.
- Glutting JJ, Youngstrom EA, Oakland T, Watkins MW (1996) Situational specificity and generality of test behaviors for samples of normal and referred children. School Psych Rev 25: 94-107.