
Journal of Metabolomics & Systems Biology
Download PDF
Research article
*Address for Correspondence: Durjoy Majumder, Department of Physiology, West Bengal State University, Berunanpukuria, Malikapur, Barasat, Kolkata 700 126, India, Email: durjoy@rocketmail.com
Citation: Chatterjee G, Mukherjee S, Majumder D. Efficacy and Toxicity Assessment of Different Clinically Used Small Molecular Tyrosine Kinase Inhibitors by Computational Molecular Docking Method. J Metabol Sys Biol. 2016;2(1): 9.
Copyright © 2016 Chatterjee G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 2, Issue: 1
Submission: 04 July, 2016 | Accepted: 26 August, 2016 | Published: 30 August, 2016
Reviewed & Approved by: Dr. Yi-Lei Zhao, Professor, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, China
Sunitinib (Sutent) is a multi-targeted receptor tyrosine kinase (TK) (>80 kinases) inhibitor that was approved by the FDA for the treatment of different cancers. Sunitinib was evaluated for its inhibitory activity against a variety of receptor protein kinases which include PDGFRa and PDGFRb; VEGFR1, VEGFR2 and VEGFR3;stem cell factor receptor (KIT), Fms-like TK-3 (FLT3), colony stimulating factor receptor Type 1 (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET) [45-47]. Sunitinib is metabolized by the liver mitochondrial CYP450 system namely CYP3A4 enzyme. Its primary active metabolite is also active. Assunitinib targets many different receptors, leads to many of its side effects. In >20% cases, the most common side effects are fatigue, asthenia, diarrhea, fever, diarrhea, nausea, mucositis/stomatitis, vomiting, dyspepsia, abdominal pain, constipation, hypertension, peripheral edema, rash, hand-foot syndrome, skin discoloration, dry skin, hair color changes, altered taste, headache, back pain, arthralgia, extremity pain, cough, dyspnea, anorexia, and bleeding. The potentially serious adverse reactions include hepato-toxicity, left ventricular dysfunction, QT interval prolongation, hemorrhage, hypertension, thyroid and adrenal dysfunction.Here attempt has been made to find out the molecular rationale of side effects and/or off-target toxicities within the physiological system by these small molecular AAG drugs.
For studying the binding of drug to protein/receptor, present work is performed with docking software Autodock 3.0 (The Scripps Research Institute, www.scripps.edu) with the help AutoDockTools (ADT), that allows the user to interact with AutoDock from a GraphicUser Inter-face (GUI) [51]. Obtained PDB files of macromolecules and ligands are further prepared by using the following steps as mentioned in the early works [52,53].

This signifies that the use of sunitinib and imatinib would be helpful in TK mediated cancer treatment management with more effective with gefitinib. The effect of gefitinib is more pronounced due to high half-life (48 hrs) (Table 1). So cancers like NSCLC, chronic phase of CML, renal cell carcinoma, advance pancreatic neuroendocrine tumor would be better managed with this drugs, but less effective in DTC, breast cancer with Her2+, advanced renal cell carcinoma,hepatocellular carcinoma. As it is less effective in metastatic breast cancer with Her2+ so, combination with capecitabine is suggested for lapatanib.

Though lapatinib is used for Her2+ breast cancer; however, our data suggested that gefitinib or sorafenib or sunitinib would be most effective (Figure 3). Probably this may be the reason that lapatinib is used in combination with capecitabine for the treatment of Her2+ breast cancer.
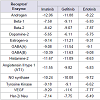
Among the studied small molecular AAG drugs (Table 3) we have found that lapatinib most strongly binds to the androgen receptor (-13.2 kcal/mol); however it has least binding capacity to the Dopamin-2 receptor (Figure 4). The highest binding energy oflaptinib to androgen receptor is more than the antagonist cyproterone to androgen receptor these signifies that use of laptinib may interfere sexual function and libido, but its effect on nausea and vomiting would be less due to less binding affinity to dopamine-2 receptor. Its binding affinity to NO synthase receptor is higher than the antagonist NNA, so it may cause cardiac dysfunction. Interestingly the binding affinity of lapatinib toward estrogen-α is high; hence it would be effective in estrogen positive breast/overian cancer (Figure 4). But due to less binding affinity of Beta-1 and Beta-2 receptor there is less chance of its effect of heart rate by this drug. It has a higher binding capacity to the GABA-A, GABA-B, histamine-2 angiotensin-II type-I nd VEGF, so the use of this drug may cause acidity sensation of pain and hypertension.

Imatinib has a high binding affinity to the androgen, histamine-2, angiotensin-II type-I and most probably similar binding affinity to GABA-A, GABA-B, NO synthase, estrogen-α, VEGF; whereas it has less binding affinity to beta-1, beta-2, dopamine-2 (Figure 5). These data signify that use of imatinib may interfere sexual function, hypertension, sensation of pain, acidity, cardiac dysfunction but heart rate, nausea, vomiting would be less due to less binding affinity to the concerned receptors.

The binding affinities of gefitinib to different receptors are in the following order: androgen < GABA-B < GABA-A < estrogen-α < histamine-2 < NO synthase < angiotensin-II type-I < beta-1 < beta-2 < dopamine-2 (Table 3). So gefitinib binds most strongly with androgen receptor while moderately binds with estrogen-α receptor (Figure 6). So it may cause anaphylaxis and lung problems, chest pain, cardiac disease, eye pain but due to less binding affinity to dopamin-2, nausea, vomiting would be less. Erlotinib has also less binding affinity to dopamine-2, so it may less effect on nausea and vomiting but it would be effect on cardiac dysfunction due to high binding affinity to the NO Synthase (Figure 7).

The binding affinity of sorafenib is high to GABA-A and GABA-B receptors and less binding affinity to beta-2 and dopamine-2 receptor, so it can cause sensation of pain, acidity and cardiac problem but nausea and vomiting hardly occurs.Sunitinib has high binding affinity to the estrogen-α and less binding affinity to beta-1 (Figure 8). This signifies it would be effective in estrogen receptor positive (ER+) breast/ovarian cancer but lesser effect on heart rate.

Efficacy and Toxicity Assessment of Different Clinically Used Small Molecular Tyrosine Kinase Inhibitors by Computational Molecular Docking Method
Gopa Chatterjee1,2, Sayan Mukherjee1 and Durjoy Majumder1,2*
- 1Department of Physiology, West Bengal State University, Berunanpukuria, Malikapur, Barasat, Kolkata 700 126, India
- 2Society for Systems Biology & Translational Research, 103 Bangur Avenue, Block - C, Kolkata 700 055, India
*Address for Correspondence: Durjoy Majumder, Department of Physiology, West Bengal State University, Berunanpukuria, Malikapur, Barasat, Kolkata 700 126, India, Email: durjoy@rocketmail.com
Citation: Chatterjee G, Mukherjee S, Majumder D. Efficacy and Toxicity Assessment of Different Clinically Used Small Molecular Tyrosine Kinase Inhibitors by Computational Molecular Docking Method. J Metabol Sys Biol. 2016;2(1): 9.
Copyright © 2016 Chatterjee G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 2, Issue: 1
Submission: 04 July, 2016 | Accepted: 26 August, 2016 | Published: 30 August, 2016
Reviewed & Approved by: Dr. Yi-Lei Zhao, Professor, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, China
Abstract
Along with the antibody based antiangiogenic (AAG) drugs, several small molecular antiangiogenic (sAAG) drugs are also in clinical practice for the treatment of different cancers. Though population based clinical trials have provided their efficacy; however, comparative efficacies of these drugs at molecular level are not available. The drug-target network is highly important to understand the efficacy and toxicity of drugs in terms of systems biology. In this study, multiply molecular docking between several sAAG drugs and different target receptors are carried out. Our data suggest that though gefitinib is quite potent in targeting different receptors involved in angigiogenesis, but it may impose some serious side effects within physiological system. Our data also suggest that one sAAG drug have some preferential binding affinity to one receptor while have lower binding capacity for other receptor. Hence different sAAG drugs may produce different types of toxicity within the physiological system. For example, lapatinib has highest binding affinity towards NO synthease, while erlotinib has lowest binding affinity to this enzyme. Similarly, sunitinib has highest binding affinity towards beta-2 receptor, while lapatinib has lowest affinity to this receptor. Hence this work provides the rationality of switching between different sAAG drugs during the course of treatment in the control of toxicity by one sAAG drug without discontinuation of therapy and management of toxicity by other drugs.Introduction
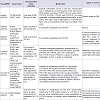
Though angiogenesis is a physiological process; however almost all types of solid tumors survive by utilizing this process. Hence, development of different therapeutic strategies that can control of tumor angiogenesis becomes an important aspect in clinical oncology [1,2]. For several types of cancer, different antiangiogenic (AAG) drugs are generally used as an adjuvant therapy that is, in combination with the conventional MTD (maximal tolerable dosing strategy) chemotherapy. Such applications have shown positiveresults with reduced (chemotherapy related) toxicities [3]. Therapy with different AAG drugs are not only supplement the ongoing chemotherapy (with docetaxel, platinum based therapies, paclitaxel, vinorelbine and gemcitabine) but also becomes important when the conventional chemotherapies have failed or are not tolerated [4]. Generally different AAG drugs are targeted towards receptors that are involved with tyrosine kinase (TK) mediated cell proliferation [5-7].Though different antibody based AAG drugs have developed and are in clinical use; however, development of different small molecular drugs that act as AAG becomes the first hand choice for cancer therapy in clinics [8,9]. There are several advantages of using small molecular AAG (sAAG) drug over antibody based AAG drugs. These drugs are more thermo-stable and can be stored without constantrefrigeration, and do not agglutinate or precipitate. They may have multiple targets, inhibit multiple kinases and target many growth factors to inhibit angiogenesis. Reports suggest that most of the small molecular tyrosine kinase inhibitors have an immunomodulatory role [10]. Perhaps the greatest advantage of sAAG drugs in future could be used for targeted therapies by inserting in liposomes [11] or can be applied through MCT (metronomic dosing strategy, where chemotherapeutic drugs are applied with dose-dense mode i.e., very low doses but with a frequent intervals) strategy; hence can be applied orally [12-18]. There are several sAAG drugs now available for clinical use and the list is growing in number; however, most of them have a molecular weight ranges from 130 to 600.
Among different sAAG drug, Imatinib mesylate (STI 571; Gleevec) was the first discovered sAAG drug that has clinical efficacy. It inhibits the Bcr-Abl tyrosine kinase (TK), the constitutive abnormal TK created by the Philadelphia chromosome t(9;22) (q34;q11) abnormality in chronic myeloid leukemia (CML). Imatinibalso inhibits the receptor tyrosine kinases for platelet derived growth factor (PDGF) and stem cell factor (SCF), called c-kit mediated cellular events (Table 1) [6,19-23]. A number of studies prove theantiangiogenic potential of imatinib. Imatinib reduces the angiogenic activity of both sporadic and neurofibromatosis Type 2 (NF2) associated vestibular schwannomas (VS) [24]. The finding is that NRP1 (Neuropilin-1) regulates angiogenesis in a VEGF (vascular endothelial growth factor) and VEGFR2-independent fashion via ABL1; hence this suggests that ABL1 inhibition provides a novel opportunity for anti-angiogenic therapy to complement VEGF or VEGFR2 blockade in eye disease [25]. Imatinib metabolism occurs in the liver mitochondria and is mediated by several isozymes of the cytochrome (CYP) P450 system, including CYP3A4 and, to a lesser extent, CYP1A2, CYP2D6, CYP2C9, and CYP2C19. The main metabolite, N-demethylated piperazine derivative, is also active. The major route of elimination is in the bile and feces; only a small portion of the drug is excreted in the urine [21,26]. A variety of adverse reactions represent local or general fluid retention including pleural effusion, ascites, pulmonary edema and rapid weight gain with or without superficial edema.
Gefitinib (Iressa) is the first discovered Epithermal Growth Factor Receptor (EGFR) associated TK inhibitor (which is also referred to as Her1 or ErbB-1). It binds to the adenosine triphosphate (ATP)-binding site of the enzyme [6,27]. Gefitinib was approved in 2003 for patients with non-small cell lung cancer (NSCLC). Theapproved indication was for the treatment of patients who were refractory to established cancer treatments (both a platinum drug and docetaxel) (Table 1) [28]. EGFR is overexpressed in the cells of certain types of human carcinomas - for example in lung and breast cancers. Over-expression of EGFR leads to inappropriate activation of the apoptotic Ras signal transduction cascade that eventually leading to uncontrolled cell proliferation. So application of Gefitinib activates the anti-apoptotic Ras signal transduction cascade in malignant cells [29]. It is primarily metabolized by CYP3A4 [30]. Side effects includeanaphylaxis (including difficulty in breathing; closing of the throat; swelling of the lips, tongue, face or hives); lung problems (difficulty breathing, shortness of breath, increased coughing, fever, or chest pain, interstitial lung disease); severe or persistent nausea, vomiting, loss of appetite, or diarrhea to grade 3 requiring interruption oftherapy for 14 days and then reinstating 250 or 500 mg dose; eyepain or irritation. Other common adverse effects include anorexia, stomatitis, dehydration, skin reactions, paronychia, asymptomatic elevations of liver enzymes, asthenia, conjunctivitis, blepharitis [31].
Erlotinib (OSI-774; Tarceva) is a small-molecule, orally dosed, anti-cancer drug that targets the EGFR (epidermal growth factor receptor) associated TK. It blocks TK mediated cell proliferation at nanomolar concentration and blocks cell cycle progression in the G1 phase. It is approved by FDA in 2004. Randomized, controlled clinical studies have demonstrated that erlotinib significantly improved survival of NSCLC patients after failure of at least one chemotherapy regimen with previously treated (with other chemotherapy) and in patients with untreated pancreatic cancer (Table 1) [6,21,32]. Erlotinib has recently been shown to be a potent inhibitor of JAK2V617F, a mutant of TK activity. JAK2V617F is found in most patients with polycythemia vera (PV) and a substantial proportion of patients with idiopathic myelofibrosis or essential thrombocythemia. The study suggests that erlotinib may be used for treatment of JAK2V617F-positive PV and other myeloproliferative disorders [33]. It is primarily metabolized by CYP3A4. It’s common side effects are rash, diarrhea, dyspnea, dry skin, back pain, chest pain, conjunctivitis, mucosal inflammation, pruritus, paronychia, arthralgia, musculoskeletal pain [34].
Lapatinib (Tykerb) is a member of the 4-anilinoquinazoline class of kinase inhibitors. It binds to human EGFR type-1 (HER1/ EGFR/ERBB1) and type-2 (HER2/ERBB2) and prevent receptorautophosphorylation upon ligand binding by TKs [35]. In combination with capecitabine, it is effective for the treatment of patients with advanced or metastatic breast cancer with overexpress HER2+ and who have received prior therapy including an anthracycline, a taxane, and trastuzumab (Table 1) [36]. At a clinically relevant concentration, lapatinib is a substrate and inhibitor of the transporter enzymes ABCG2 or BCRP (a wild type Breast Cancer Resistance Protein), P-gp (or ABCB1, ATP Binding Cassette subfamily B member 1) and also an inhibitor of OATP1B1 (Organic Anion Transporter Pump family 1B1) or Solute carrier organic anion transporter family member 1B1 (SLCO1B1) [37]. Lapatinib undergoes metabolism, predominantly by liver mitochondrial CYP450 enzyme system specially CYP3A4 and CYP3A5, with minor contributions from CYP2C19 and CYP2C8 to a variety of oxidized metabolites, to form a variety of metabolites. Side effects of lapatinib include hepatotoxicity (increase in liver enzymes and cholestatic liver damage by disrupting of hepatocyte bile salt efflux and a decreased of left ventricular ejection fraction (LVEF). 10% patients experience diarrhea, rash, nausea and fatigue when lapatinib is applied in combination with letrozole [38-41]. Lapatinib has also been associated with interstitial lung disease and pneumonitis in monotherapy or in combination with other chemotherapies [38].
Sorafenib (Nexavar, BAY 43-9006) is unique in targeting the Raf/Mek/Erk pathway. By inhibiting these kinases, genetic transcription involving cell proliferation and angiogenesis is inhibited. However it also inhibit a variety of kinase receptors, including VEGFR, EGFR,and PDGFR kinases; so it is recommended for different types of cancer (Table 1) [6,42,43] Sorafenib is metabolized primarily by CYP3A4 as well as glucuronidation (mediated by UGT1A9): 77% of the dose excreted in feces and 19% of the dose excreted in urine as glucuronidated metabolites. At steady state the metabolites of sorafenib are detected in plasma. Pyridine N-oxide, the main circulating metabolite of sorafenib in plasma, shows in vitro potency similar to that of sorafenib. Side effects include cardiac ischemia, infarction, hypertension, hemorrhage, hand-foot skin reaction, rash, Stevens-Johnson syndrome, and toxic epidermal necrolysis, gastrointestinal perforation, QT interval prolongation, drug induced hepatitis, hypoalbuminemia, elevated amylase, thrombocytopenia, hypocalcaemia [44].
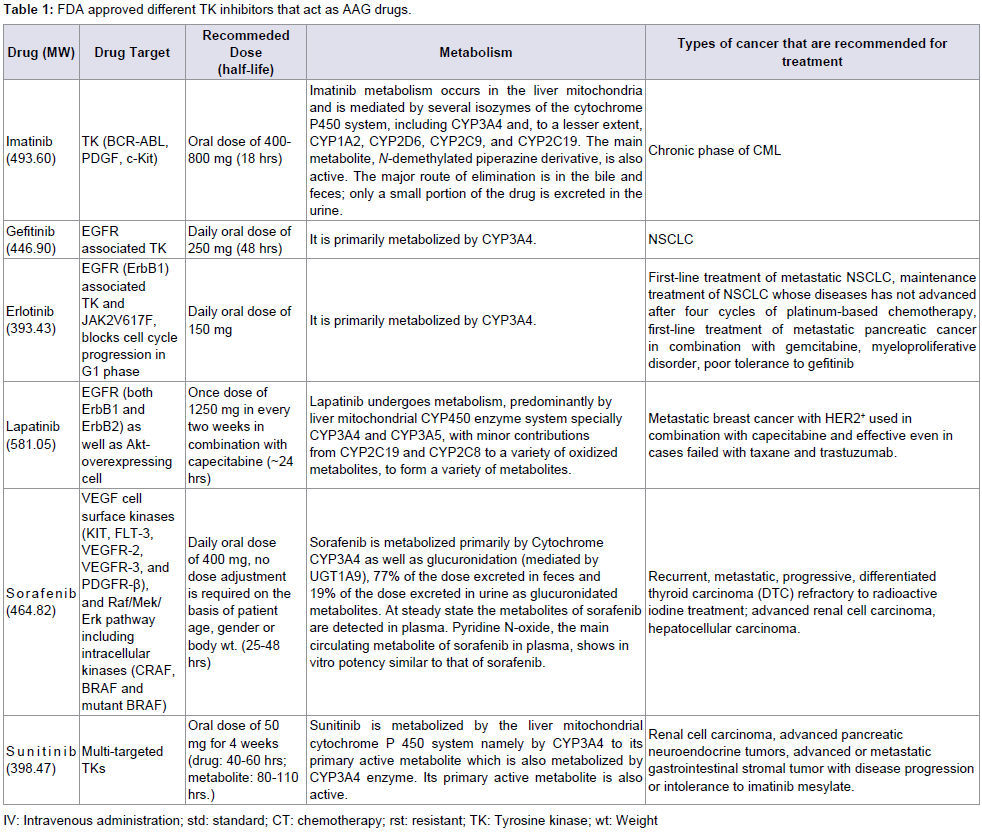
Table 1: FDA approved different TK inhibitors that act as AAG drugs.
Materials and Methods
All data used in the present work are collected from the data resources that are available in the public domain and majority of the used data and its resources are already mentioned in an early work [48]; however, the newly added data (along with its resources) are mentioned in Table 2. The protein/macromolecular 3D structuraldata and the 3D drug structures are downloaded from the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do) [49] and the Drug Bank (http://redpoll.pharmacy.ualberta.ca/drugbank/) database [50].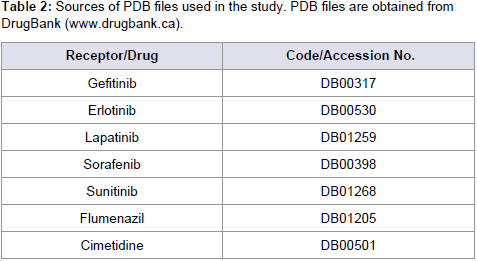
Table 2: Sources of PDB files used in the study. PDB files are obtained fromDrugBank (www.drugbank.ca).
1. Preparing the macromolecular PDB files - as the PDB files often contained added waters, these water molecules were files were selected as HOH* from a string and deleted after the warning. Polar hydrogens with no bond order and Kollman charges were added [bydefault; ADT adds Kollman charges for a peptide (determined by checking whether all of its component residue names appear in the standard set of 20 commonly occurring amino acids) and Gasteiger charges if not so]. Finally, solvation parameters were added and the files saved as molecule. pdbqs (where “q” and “s” represent charge and solvation, respectively).
2. Preparation of the ligand file - Generally all hydrogens are added and non-polar hydrogens are merged. Gasteiger charges added unless the ligand is also a peptide in which case the above procedurewould be followed. ADT automatically takes care of solvation and checks for aromatic carbon atoms and hence lone pairs and non polar hydrogens merged. ADT then determined the best root (the best root is the atom in the ligand with the smallest or largest sub-tree; in case of a tie, if either atom is in a cycle, it is picked as the root, and if neither or both is in a cycle, the first found is picked). Next we defined rotatable bonds in the ligand, making all amide bonds non-rotatable, and set the number of active torsions to fewest atoms. The ligand filewas then saved with a ligand.out.pdbq extension (“q” representing charge).
3. Preparation of the grid and the grid parameter file - For the calculation of docking interaction energy, 3D grid box was created in which the protein molecule was enclosed. The grid volume was set to be large enough to allow the ligand to rotate freely; even with it’s full extended conformation. The parameters required to create such agrid were stored in the grid parameter file, molecule.gpf.
4. Now autogrid3 was run to create a map for every atom type in the ligand and create the corresponding macromolecular file with the extension molecule.glg either from Run of the GUI or the commandline autogrid3 -p molecule.gpf -l molecule.glg and when finished it writes Successful Completion.
5. Preparation of Docking Parameter File - The macromolecular pdbqs and ligand.out.pdbq files were read. The search methods of AutoDock include the Monte Carlo simulated annealing method, thegenetic algorithm, local search, and the hybrid genetic algorithm with local search. The latter is also referred to as the Lamarckian genetic algorithm because offspring’s are allowed to inherit the local search adaptations of their parents, and this was the chosen algorithm for the analysis. The docking job can similarly be run from Run of the GUI or from command line autodock3 -p molecule.dpf -l molecule.dlg and when finished, Successful Completion was written the PDB file and Analyze open docking log is done and choosing different color for receptor and ligand the conformations could be played by energy. The dlg files can be opened in a terminal and each run’s (the number of runs can be fixed by the user) final docked energy, Gibbs free energy, Inhibition Constant is written. Also given in the file are the RMSDvalues. A conformation can be chosen and it’s coordinates writtento run the next set of docking and this should be done till Gibb’s free energy is no longer significantly reduced.
6. Diagrams were drawn with the protein and the ligand attached to it in its lowest energy conformer. Protein and ligand can be made of different width and color and the background white. The image can be saved in various formats of which we chose the tif format.
Results
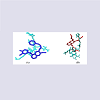
Rigorous docking experiments had been performed to assess the comparative efficacy between clinically used TK inhibitors (drugs) and their cross-reactivity to different receptors and/or enzymes ithin the physiological system. The TK drugs, the receptors, and the corresponding docking results are listed in Table 3.Among the studied small molecular AAG drugs we have found that gefitinib most strongly binds to the VEGF receptor (-11.6 kcal/mol); whereas erlotinib has less binding affinity to the VEGF (Table 3 and Figure 1). However, sunitinib & imatinib have high binding affinity to the VEGF; whereas lapatinib and sorafenib have less binding affinity.
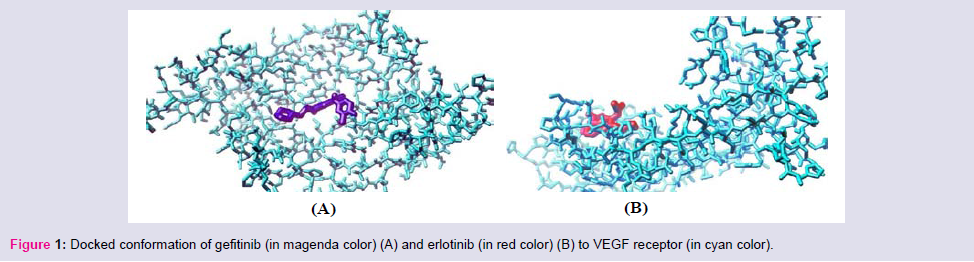
Figure 1: Docked conformation of gefitinib (in magenda color) (A) and erlotinib (in red color) (B) to VEGF receptor (in cyan color).
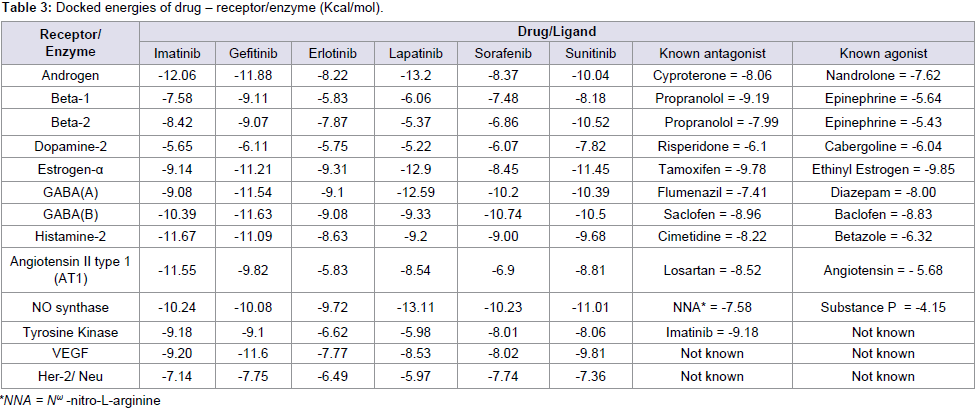
Table 3: Docked energies of drug – receptor/enzyme (Kcal/mol).
Among the studied drugs our results indicate that binding affinities of the drugs to the TK receptor are in following order: imatinib ≤ gefitinib < sunitinib < soratinib < erlotinib < lapatinib. So imatinib has the highest binding affinity to TK receptor; hence it may be the ideal choice for treatment of CML (Figure 2). Gefitinib has almost the same binding affinity, hence may be recommended for others myeloproliferative disorder and NSCLC. Likewise sunitinib and sorafenib are recommended for different epithelial cancers with recurrent metastatic, or progressive cancers of thyroid, kidney, liver and GI system.
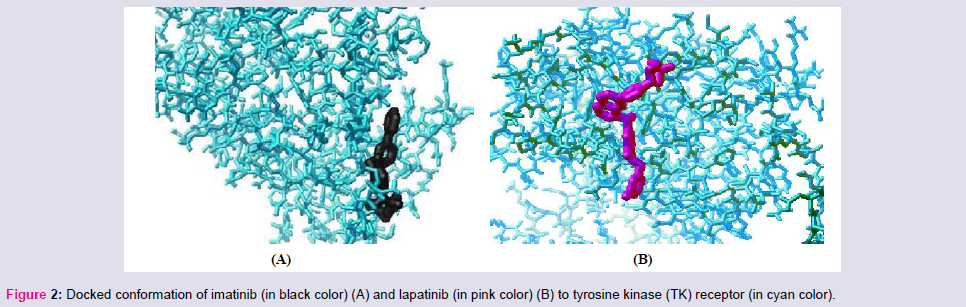
Figure 2: Docked conformation of imatinib (in black color) (A) and lapatinib (in pink color) (B) to tyrosine kinase (TK) receptor (in cyan color).

Figure 3: Docked conformation of sorafenib (in violet color) (A) and sunitinib (in blue color) (B) to Her-2 receptor (in cyan color).
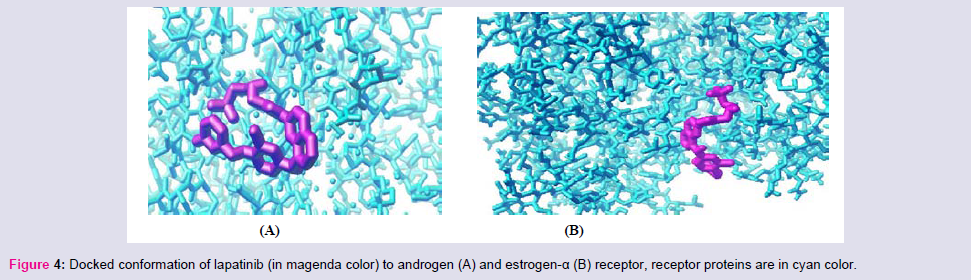
Figure 4: Docked conformation of lapatinib (in magenda color) to androgen (A) and estrogen-α (B) receptor, receptor proteins are in cyan color.
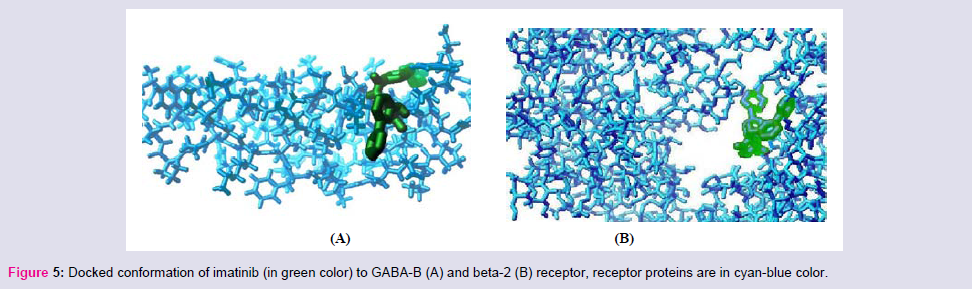
Figure 5: Docked conformation of imatinib (in green color) to GABA-B (A) and beta-2 (B) receptor, receptor proteins are in cyan-blue color.

Figure 6: Docked conformation of gefitinib (in blue color) to androgen (A) and estrogen-α (B) receptor, receptor proteins are in cyan color.
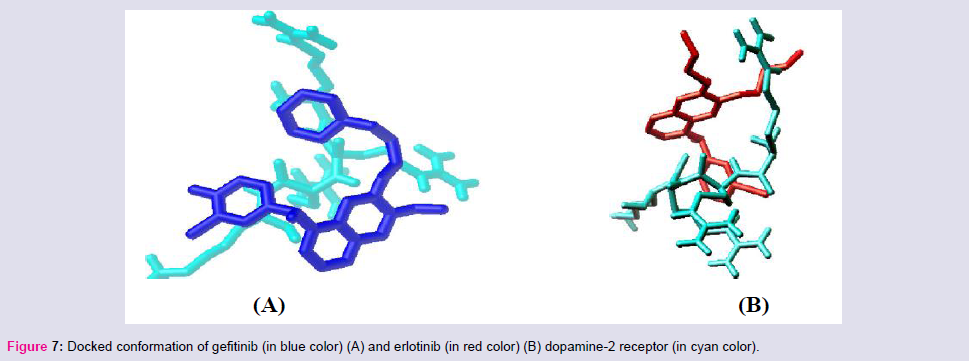
Figure 7: Docked conformation of gefitinib (in blue color) (A) and erlotinib (in red color) (B) dopamine-2 receptor (in cyan color).
Figure 8: Docked conformation of sunitinib (in blue color) to estrogen-α (A) and beta-1 (B) receptor, receptor proteins are in cyan color
Discussion
To circumvent the toxic side effect of conventional chemotherapy as well as to make better control over tumor growth different antibody based AAG drugs were developed. Primarily Bevacizumab and trastuzumab were developed; both of them are now in clinical use. Though it was apprehended that they don’t possess any toxic side effect but clinical data suggest that these drugs have toxic side effects of the physiological system. We have performed Molecular docking experiment between these drugs and array of receptors and enzyme molecules of the physiological system, our data supports the clinical findings of toxicity by these drugs [48].For clinical feasibility like storage and application to patients through different routes, different small molecular drugs have also been developed. Though several empirical based experimental as well as clinical data are available regarding their efficacy, however no comparative data in quantitative terms are available. Here we have performed computational docking method to make an assessment regarding their efficacy in quantitative terms for small molecular AAG drugs. We have found that binding efficacy of Imatinib (to TK receptor) is most potent. Gefitinib binds to TK receptor almost the same efficacy with imatinib; however, binding capacity of gefitinib to VEGF, Her-2 is the most potent. Among the studied small molecular AAG drugs; however, lapatinib has least binding affinity to TK receptor and Her-2 and erlotinib has least binding affinity to VEGF receptor. The overall finding of this work suggests that gefitinib is the most potent broad spectrum sAAG drug.
Similar to the antibody based AAG drugs several reports are available which indicate that these small molecular AAG drugs also have some toxic side effects. This may be due to cross-reactivity of these drugs to the physiological system. Previously it has been shown that different small molecular chemotherapeutic drugs when applied on a long term basis may produce off target binding ability and henceproduce toxic side effect [52] and computational docking method could be the way out to prove this in a quantitative terms so that offtarget oints can be graded.
Off target toxicity of a drug can be assessed if that particular drug binds to a receptor with the same binding efficacy compare to its known antagonist/agonist. Though gefitinib can be regarded as the potent small molecular AAG drug, however its binding capacity is either more or almost same to the known antagonist/agonist of the studied receptor/enzyme molecules. Imatinib may has strong cross reactivity to almost all the studied receptor/enzyme namely androgen, beta-2, estrogen-α, GABA-B receptor, histamine, angiotensin-II type-I receptor and NO Synthase. Though sunitinib can be consider as an effective sAAG drug after gefitinib but it has also very high binding capacity to beta-2, dopamin-2, estrogen-α,GABA-B receptors. Lapatinib has intermediate binding affinity to VEGF receptor but it has a strong binding affinity to GABA-A receptor. Our data suggest that erlotinib has less affinity to receptors involved in angiogenesis and also less toxicity potential compared to other small molecular AAG drugs. Interestingly though sorafenib has lesser binding affinity compared to gefitinib to TK, VEGF and Her-2, and it has also very lesser amount of binding affinity to the studied physiological receptor. Hence, this study would help in rationalizing to switch over from one drug to another during the intermittent period of toxicity, thereby discontinuation of therapy or adoption of other drug for management may be prevented. While make an attempt to study the off target toxicity an interesting finding may beconcluded from our study is that these small molecular AAG drugs can be used for estrogen positive breast/overian cancer.
Androgen receptor is most effected by lapatinib and then imatinib < gefitinib < sunitinib < sorafenib < erlotinib in a sequential manner. So this result may hind toward the cause of fatigue by the use of laptinib. Beta-1 receptor is mostly affected by gefitinib while beta-2 receptor mostly affected by sunitinib; however, almost sameeffects were noted for other drugs. This finding indicates cardiac dysfunction, ventricular dysfunction, pulmonary hypertension and peripheral edema. Dopamine-2 receptor is mostly affected by sunitinib also and all other drugs with almost same efficiency. This indicate that why nausea and vomiting occurs during application of small molecular AAG drugs. Lapatinib, sunitinib, gefitinib affects estrogen-α in the almost same efficiency. This may indicate the cause of fatigue, cholestasis liver damage, hepatocyte damage, dyspnea, adrenal dysfunction, anaphylaxis reaction, breathing problem that occurs after application by these drugs. When gefitinib is used, GABA receptor (both GABA-A and GABA-B) are affected mostly, this hind toward the cause of interstitial lung disease with chest pain by the use of this drug, similar inference can be drawn with Lapatinib. Histamin-2 receptor is mostly affected by imatinib and gefitinib followed by sunitinib, lapatinib, sorafenib, erlotinib and binding efficiency of all of these drugs are much more to their antagonist/agonist, these indicate that why anorexia, nausea, loss of appetite is commonly observed by the application of any of these drugs. Binding to angiotensin-II type-I receptor of the drugs are in the order: imatinib< genitinib < sunitinib < lapatinib < sorafenib and majority of them; and binding energy all of these drugs are more than their agonists. Therefore it may be concluded that why majority of the drugs causes ither hypertension or myocardial infarction. By our study it is noted that most of the sAAG drugs bind efficiency to the NO synthase, compare to the binding efficiency of their corresponding antagonist/ agonist. Though cardiac dysfunction, myocardial infarction are notable side effect in sorafenib treatment; however, in general fatigue, hypertension and peripheral edema are seen as side effect during treatment with lapatinib, imatinib and sunitinib may be due to the effect of these drugs on NO synthase.
The drug-target network is highly important to understand the efficacy and toxicity in terms of systems biology. This study may help in an understanding to take precautionary steps to prevent the toxic side effects; if possible, different dynamical models are also developed to pin-point the onset of toxicity for long term useof chemotherapeutic drugs. Hence the grading approach of organs/ tissue specific toxicity may provide strong impetus to the dynamical model in the assessment of very onset of development by any particular drug. This way the observed efficacy and toxicity from this study would be fully understandable. Hence to explore the off-target toxicities by these small molecular AAG drugs has of relevance.
Acknowledgements
GC and SM contributed equally to this work. Authors acknowledge the critical comments of SSBTR (Society for Systems Biology & Translational Research) members on this manuscript.References
- Ausprunk DH, Folkman J (1977) Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumour angiogenesis. Microvasc Res 14: 53-65.
- Risau W (1997) Mechanisms of angiogenesis. Nature 386: 671-674.
- Lee JM, Sarosy GA, Annunziata CM, Azad N, Minasian L, et al. (2010) Combination therapy: intermittent sorafenib with bevacizumab yields activity and decreased toxicity. Br J Cancer 102: 495-499.
- Schettino C, Bareschino MA, Ricci Ciardiello F (2008) Erlotinib: an EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev Respir Med 2:167-178.
- Kitano H (2002) Kim DW, Lu B, Hallahan DE (2004) Receptor tyrosine kinase inhibitors as anti-angiogenic agents. Curr Opin Investig Drugs 5: 597-604.
- Arora A, Scholar EM (2005) Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 315: 971-979.
- Jeltsch M, Leppanen VM, Saharinen P, Alitalo K (2013) Receptor tyrosine kinase-mediated angiogenesis, Cold Spring Harb Perspect Biol 5: a009183.
- Wu H-C, Huang CT, Chang DK (2008) Anti-angiogenic therapeutic drugs for treatment of human cancer. J Cancer Mol 4: 37-45.
- Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression and metastasis. Nat Rev Clin Oncol 8: 210-221.
- Heine A, Held SA, Bringmann A, Holderried TA, Brossart P (2011) Immunomodulatory effects of anti-angiogenic drugs. Leukemia 25: 899-905.
- Krasnici S, Werner A, Eichhorn ME, Schmitt-Sody M, Pahernik SA, et al. (2003) Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer 105: 561-567.
- Majumder D, Mukherjee A (2007) Mathematical modeling of toxicity-related trade-offs in metronomic chemotherapy. IET Syst Biol 1: 298-305.
- Mukherjee A, Majumder D (2008) Mathematical modelling for the assessment of the effect of drug application delays in metronomic chemotherapy of cancer due to physiological constraints. Biosystems 91: 108-116.14.
- Majumder D (2010) Tumor angiogenesis based analytical model for the assessment of MCT and MTD chemotherapeutic strategies in cancer. J Biol Syst 18: 749-761.
- Mukherjee A, Majumder D (2010) Dynamical model for the assessment of anti-angiogenic therapy of cancer. Mol Biosyst 6: 1047-1055.
- Majumder D, Mukherjee A (2011) A passage through systems biology to systems medicine: adoption of middle-out rational approaches towards the understanding of therapeutic outcomes in cancer. Analyst 136: 663-678.
- Dhar PK, Majumder D (2013) A composite synergistic systems model for exploring the efficacies of different chemotherapeutic strategies in cancer. Comput Biol J 1-26.
- Majumder D, Mukherjee A (2013) Multi-scale modeling approaches in systems biology towards the assessment of cancer treatment dynamics: adoption of middle-out rationalist approach. Adv Cancer Res Treat.
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, et al. (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2: 561-566.
- Morin MJ (2000) From oncogene to drug: development of small molecule tyrosine kinase inhibitors as anti-tumor and anti angiogenic agents. Oncogene 19: 6574-6583.21.
- (2008) Highlights of prescribing information. US Food and Drug Administration.
- Okada M, Adachi S, Imai T, Watanabe K, Toyokuni SY (2004) A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood 103: 2299-2307.
- Fausel C (2007) Targeted chronic myeloid leukemia therapy: seeking a cure. Am J Health Syst Pharm 64(24 Suppl 15): S9-S15.
- Raimondi C, Fantin A, Lampropoulou A, Denti L, Chikh A, et al. (2014) Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J Exp Med 211: 1167-1183.
- Stegmeier F, Warmuth M, Sellers WR, Dorsch M (2010) Targeted cancer therapies in the twenty-first century: lessons from imatinib. Clin Pharmacol Ther 87: 543-552.
- Gschwind HP, Pfaar U, Waldmeier F, Zollinger M, Sayer C, et al. (2005) Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos 33: 1503-1512.
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, et al. (2004) EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 101: 13306-13311.
- FDA U.S. Food and Drug Administration (2005) Information for healthcare professionals: Gefitinib (marketed as Iressa).
- Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, et al. (2000) Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 6: 2053-2063.
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129-2139.
- Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, et al. (2010) Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 11: 121-128.
- Rocha-Lima CM, Raez LE (2010) Erlotinib (tarceva) for the treatment of non-small-cell lung cancer and pancreatic cancer. Clin Cancer Res 16: 1673-1681.
- Li Z, Xu M, Xing S, Ho WT, Ishii T, et al. (2007) Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J Biol Chem 282: 3428-3432.
- Tevaarwerk AJ, Kolesar JM (2009) Lapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin Ther 31 Pt 2: 2332-2348.
- (2013) Highlights of prescribing information. Astellas Pharma US.
- Burris HA 3rd, Hurwitz HI, Dees EC, Dowlati A, Blackwell KL, et al. (2005) Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol 23: 5305-5313.
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O'Mara MJ, et al. (2008) The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 36: 695-701.
- (2013) Highlights of prescribing information.
- Teng WC, Oh JW, New LS, Wahlin MD, Nelson SD, et al. (2010) Mechanism-based inactivation of cytochrome P450 3A4 by lapatinib. Mol Pharmacol 78: 693-703.
- Castellino S, O'Mara M, Koch K, Borts DJ, Bowers GD, et al. (2012) Human metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity. Drug Metab Dispos 40: 139-150.41.
- Pauli-Magnus C, Meier PJ (2006) Hepatobiliary transporters and drug-induced cholestasis. Hepatology 44: 778-787.
- BAX: ligand summary. Protein Data Bank.
- Kim JE, Ryoo BY, Ryu MH, Chang HM, Suh DJ, et al. (2012) Sorafenib dose escalation in the treatment of advanced hepatocellular carcinoma. Oncology 82: 119-125.
- (2005) Highlights of prescribing information. Onyx Pharmaceuticals.
- Sutent (2011) Highlights of prescribing information. Pfizer Labs.
- B49: ligand summary. Protein Data Bank.
- 47. Roskoski R Jr (2007) Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun 356: 323-328.
- Mukherjee S, Chatterjee G, Ghosh M, Das B, Majumder D (2016) Efficacy and toxicity assessment of different antibody based antiangiogenic drugs by computational docking method. Adv Bioinformatics 2016: 7053712.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. (2000) The protein data bank. Nucleic Acids Res 28: 235-242.
- Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, et al. (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34 (Database issue): D668-D672.
- Huey R, Morris GM (2006) Using autodock with autodocktools: a tutorial. The Scripps Research Institute, Molecular Graphics Laboratory, USA 1-51.
- Mukherjee S, Majumder D (2009) Computational molecular docking assessment of hormone receptor adjuvant drugs: breast cancer as an example. Pathophysiology 16: 19-29.
- Majumder D, Mukherjee S (2013) Molecular docking assessment of efficacy of different clinically used arsenic chelator drugs. J Comput Med 2013: 1-8.