
Research Article
*Address for Correspondence: Anelia Horvath, McCormick Genomics and Proteomics Center, Department of Biochemistry and Molecular Medicine, The George Washington University, USA Tel: +1 (202)-994-2114; Fax: +1 (202)-994-8974; E-mail: horvatha@gwu.edu
Citation: Mudvari P, Kowsari K, Cole C, Mazumder R, Horvath A. Extraction of Molecular Features through Exome to Transcriptome Alignment. J MetabolSys Biol. 2013;1(1): 7.
Copyright © 2013 Anelia Horvath et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 1, Issue: 1
Submission: 30 June 2013 | Accepted: 20 August 2013 | Published: 22 August 2013


Overlay of the four datasets revealed several intriguing observations (Figure 1A). First, both normal and tumor transcriptome displayed more SNPs as compared to the corresponding exomes (Table 2). More than 70% of the variants seen in the transcriptome and not in the exome overlapped between the normal and the tumor datasets. When we analyzed the variants by annotation type, we found that the major proportion of the transcriptome exclusive variants (78% and 67%, for the normal and the tumor, respectively) belong to 3’ and 5’UTRs. Such a result is logical due to the widely used exome capture design that includes only the UTR regions immediate to the coding sequences. Therefore, most of the UTR variants called are outside the exome capture target. In contrast, since these regions are transcribed, they are readily included in the transcriptome and thus comprise the major variant-based difference between the exome and the transcriptome.

From the remaining, at least half are called with 5 or less reads, with some proportion of those likely to represent false positives, especially the ones not listed in the human variation databases (referred hereafter as “novel”). A portion of the non-UTR transcriptome-exclusive variants likely reside outside of the exome capture capacity and, finally, a small percentage may represent post-transcriptional modifications, including RNA editing events.


In the tumor setting, a common cause for allelic expression is the elimination of one of the alleles, in many cases the wild type, through the mechanism of LOH. Because LOH occurs at DNA level, such SNPs can be identified by their homozygote vs heterozygote state in the tumor and the normal exomes, respectively. In our dataset, 214 SNPs, five of which are novel, matched these conditions. Since LOH usually affects large genomic regions, one additional distinguishing feature would be the coexistence, in an uninterrupted fashion, of similarly transitioned hetero-to-homozygote SNPs (from normal to tumor exome, respectively) in the immediate chromosomal surroundings. Several strings of adjacently located SNPs wereobserved in our dataset; an example is the gene string TNS1, PNKD, ZNF142, and OBSL1, encompassing the region chr2:218682771 - chr2:220431631 (rs1043537 is presented on Figure 2C). Of note, somatic deletion of these genes was confirmed by the microarray data available on the same sample through the integrative cBioPortal for Cancer Genomics (http://www.cbioportal.-org/public-portal/). This validation shows the capacity of the exome-to-transcriptome alignment to independently indicate potential LOH, and, at the same time, features one additional level of genomic data integration.
Despite the array of integrative NGS analyses provided advantages, on many occasions they need to be applied with caution. Here, we highlight three important points worthy of consideration when aligning NGS datasets. First, ample coverage for all compared regions is necessary, to avoid methodological bias. While in many cases low number of RNA-seq reads indicate low expression, it may also reflect difficult to sequence transcripts. This consideration further applies on preferentially expressed variants - either the nucleotide change itself, or, coexisting allelic feature can provide sequencing advantage or disadvantage compared to the wild type. In both cases, alternative method and/or multi-samples comparisons are the first step to confirm the authenticity of the observation.
Extraction of Molecular Features through Exome to Transcriptome Alignment
Prakriti Mudvari1, Kamran Kowsari1, Charles Cole2,Raja Mazumder2 and Anelia Horvath1,2*
- 1McCormick Genomics and Proteomics Center, USA
- 2Department of Biochemistry and Molecular Medicine, The George Washington University, Washington, District of Columbia 20037, USA
*Address for Correspondence: Anelia Horvath, McCormick Genomics and Proteomics Center, Department of Biochemistry and Molecular Medicine, The George Washington University, USA Tel: +1 (202)-994-2114; Fax: +1 (202)-994-8974; E-mail: horvatha@gwu.edu
Citation: Mudvari P, Kowsari K, Cole C, Mazumder R, Horvath A. Extraction of Molecular Features through Exome to Transcriptome Alignment. J MetabolSys Biol. 2013;1(1): 7.
Copyright © 2013 Anelia Horvath et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 1, Issue: 1
Submission: 30 June 2013 | Accepted: 20 August 2013 | Published: 22 August 2013
Abstract
Integrative Next Generation Sequencing (NGS) DNA and RNA analyses have very recently become feasible, and the published to date studies have discovered critical disease implicated pathways, and diagnostic and therapeutic targets. A growing number of exomes, genomes and transcriptomes from the same individual are quickly accumulating, providing unique venues for mechanistic and regulatory features analysis, and, at the same time, requiring new exploration strategies. In this study, we have integrated variation and expression information of four NGS datasets from the same individual: normal and tumor breast exomes and transcriptomes. Focusing on SNP-centered variant allelic prevalence, we illustrate analytical algorithms that can be applied to extract or validate potential regulatory elements, such as expression or growth advantage, imprinting, loss of heterozygosity (LOH), somatic changes, and RNA editing. In addition, we point to some critical elements that might bias the output and recommend alternative measures to maximize the confidence of findings. The need for such strategies is especially recognized within the growing appreciation of the concept of systems biology: integrative exploration of genome and transcriptome features reveal mechanistic and regulatory insights that reach far beyond linear addition of the individual datasets.Keywords
Exome; Transcriptome; Breast Tumor; Breast Cancer; SNP; Allelic Imbalance; Allele Preferential Expression; RNA Editing; Somatic Mutations; Imprinting; LOHIntroduction
With the evolution of next-generation sequencing (NGS) technologies, the time, cost and amount of the material needed are constantly declining, thus making applications such as genome/exome and transcriptome sequencing increasingly feasible. As a result, a rapidly growing number of exomes, genomes and transcriptomes from the same individual are accumulating, providing unique venues for mechanistic and regulatory feature analysis, and, at the same time, requiring new exploration strategies. To date, only a handful of studies have integrated NGS genome scale datasets. Nevertheless, these studies have provided essential functional and regulatory insights, reaching far beyond linear addition of individual NGS dataset information layers, and often unraveling novel diagnostic and therapeutic targets [1-6].Commonly explored algorithms for genomic data integration include alignment of germ line and somatic DNA in search for tissue and tumor-specific changes [6], exome/genome-to-transcriptome comparison for pre- and post-transcriptional regulatory elements [7], and non-coding transcriptome-epigenome-genome overlay for assessment of encoded and acquired expression control [8-11]. In our study, we focus on one relatively unexplored aspect of integrative genomic analysis: SNP-centered allelic preferential expression at nucleotide resolution using exome and transcriptome data from the same individual.
We have integrated the variation and expression information of four NGS datasets from the same individual: germ-line exome,normal breast tissue transcriptome, and breast tumor exome and transcriptome (Table 1). Focusing on nucleotide resolution allelic imbalance, we explore different analytical algorithms to retrieve potential encoded-to-regulatory links: expression/growth advantage driving variations, tumor-related gross genomic alterations, somatic changes, imprinting, and, RNA editing. We further discuss our observations in the light of existing knowledge, and highlight opportunities to integrate expression data through variation-to-abundance analytical algorithms. Finally, we point to some critical elements that might bias the output and recommend alternative measures to maximize the confidence of the findings.
Table 1: Sample attributes of data included in our study.
Materials and Methods
Short read data was obtained from The Cancer Genome Atlas (http://cancergenome.nih.gov/) via the CGHub data portal (https://cghub.ucsc.edu/). Short read datasets from a single patient with exome and RNA-seq breast cancer tumor and normal tissue (TCGABH-A0B3) was selected for analysis. Additional information about the patient and sample was retrieved from data matrix available at http://tcgadata.nci.nih.gov/tcga/dataAccessMatrix.htm. The sample details are as follows: Disease type - BRCA-Breast invasive carcinoma; Data Type - Clinical; Race - White; History - no previous history of malignancy; Platform - illumina. The tumor and the matched control samples were identified by the TCGA bar code associated with the sample. The sample type of RNA-seq case dataset (TCGA-BH-A0B3- 01A-11R-A056-07) is Primary Tumor. The sample type of RNA-seq control datasets (TCGA-BH-A0B3-11B-21R-A089-07) is Normal Solid Tissue. The sample type of exome case datasets (TCGA-BHA0B3- 01A-11W-A071-09) is Primary Tumor. The sample type of exome control datasets (TCGA-BH-A0B3-10A-01W-A071-09) is Blood Derived Normal. The data manifest was downloaded using the ‘cgquery’ script, which is available on the CGHub website (https:// cghub.ucsc.edu/). A manifest file was generated by specifying the required characteristics, such as disease type, platform, tissue site, etc. Once the manifest was generated, data was downloaded through the ‘GeneTorrent’ software (also available on CGHub) in BAM format. The BAM file is converted into the FASTQ format using the ‘bam2fastq’ program available through http://www.hudsonalpha.org/-gsl/information/software/bam2fastqThe raw reads were then aligned against Ensembl GRCh37 (hg19) using Bowtie2 [12] for exome data and TopHat2 [13] for transcriptome data. For transcript assembly, we utilized the Cufflinks from the Tuxedo suite of programs [14]. We used the default parameters during the analysis, which filters low abundance transcripts that comprise 10% of the most abundant isoform. The variants were called using mpileup utility of Samtools [15] with default settings except for the maximum read depth, which was set to 8,000. The variation calls were annotated through SeattleSeq v.137 (http://snp.gs.washington.edu/SeattleSeqAnnotation137/). To minimize false negative and false positive calls, we applied filtering as previously described [16]. Briefly, previously reported SNPs, due to independent validation by other group(s), were analyzed further without filtering. The novel variants were required to satisfy the following criteria optimized by our group: supported by a minimum of four bidirectional reads with unique start position, phred quality value (QUAL) > 20, and mapping quality value (MQV)>20. For visual evaluation of positions of interest, we utilized Integrative Genomic Viewer (IGV, http://www.broadinstitute.org/ software-/igv/home) [16,17].
Results
Prior to integration of the datasets, all variants were called and annotated individually in the four datasets, and abundance was estimated for the transcriptomes. To extract observations with high confidence, we analyzed the above features only in regions well covered in all four datasets. To maximize the informative overlap between the exome and transcriptome, we filtered out data from intergenic and intronic regions. As an initial step for the integrative analysis, we mapped all the SNPs called in at least one dataset, and displayed the wild type (wt) and variant (var) calls in absolute and relative (var/wt) numbers for all four datasets. The major steps of our analytical algorithm are presented in (Figure 1).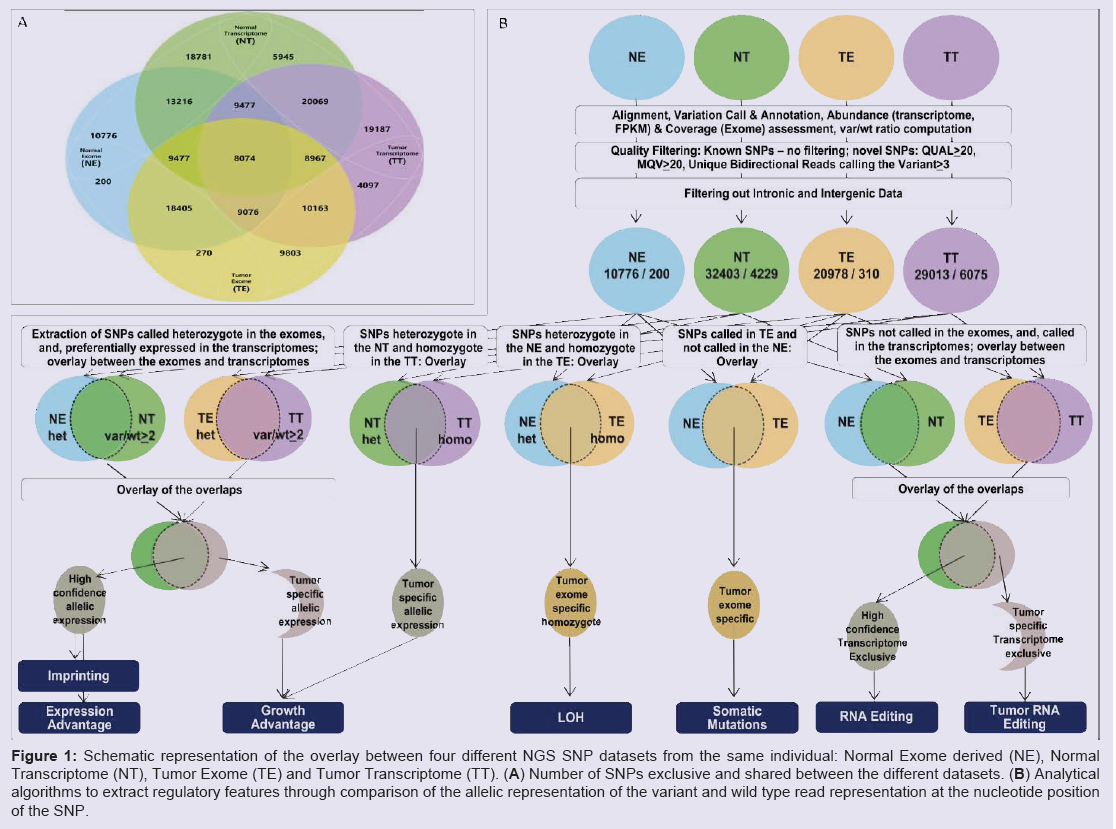
Figure 1: Schematic representation of the overlay between four different NGS SNP datasets from the same individual: Normal Exome derived (NE), Normal Transcriptome (NT), Tumor Exome (TE) and Tumor Transcriptome (TT). (A) Number of SNPs exclusive and shared between the different datasets. (B) Analytical algorithms to extract regulatory features through comparison of the allelic representation of the variant and wild type read representation at the nucleotide position of the SNP.
Table 2: Number of SNPs exclusive and shared between the four analyzed datasets.
To outline preferentially expressed SNP bearing alleles, we extracted all transcriptome SNPs called with var/wt ratio higher than 2 (var/wt > 2), and, called heterozygote in the respective exomes (Figure 1A). Overlap of thus selected normal and tumor datasets revealed higher-confidence preferentially expressed SNPs, and the ones exclusive for the tumor set defined a subset of potential tumorspecific changes. From the preferentially expressed alleles, a particular over-selected subset consists of the novel SNPs, due to the minimal possibility to be present in homozygote state on genomic level. For high confidence findings on this dataset, we further tightened the criteria for strong allele preferential expression to var/wt > 5. This revealed thirty-one SNPs, all present in the population databases, shared between the normal and the tumor datasets; seven of them were homozygous in both transcriptomes. When we looked in the tumorspecific allele-preferential expression, we found 683 SNPs with strong allele-preferential expression exclusive for the tumor transcriptome; from them, 11 were novel (not present in the databases) and 343 were in homozygous state.
Several molecular mechanisms may account for preferential expression of genomically heterozygous SNPs in the normal transcriptome. Among the common causes are imprinting, caused by the exclusive expression from only one of the parental alleles, and, cis-acting expression advantage provided by the allele harboring the SNP. In some cases this might be caused by the nucleotide change contained in the SNP of interest, through either creation or disruption of regulatory molecule(s) binding element. In our dataset, an example of imprinting is illustrated by the known SNP (rs2192206) in the gene encoding the growth suppressor necdin NDN (Table 3) NDN is known for its exclusively paternal expression [19]; rs2192206 presented with well-balanced heterozygous signal in both exomes and monozygotic expression of the variant allele in both transcriptomes (Figure 2A). In contrast, rs73231013 in the gene encoding the nucleosome binding protein HMGN5 shows similar expression pattern (Figure 2B) without acknowledged involvement of imprinting processes. Whether the SNP rs73231013 in HMGN5 is molecularly implicated in the increased allelic expression is a subject of future large-scale validation and focused wet-lab studies.
Table 3: Illustrative examples of SNPs representing different regulatory features. The chromosomal coordinates of the change, the harboring gene, the functional annotation, and the respective number of variation and wild type calls are presented for each SNP.
*Total number of reads, before filtering for MQV; reads will MQV< 20 were removed from further analysis.
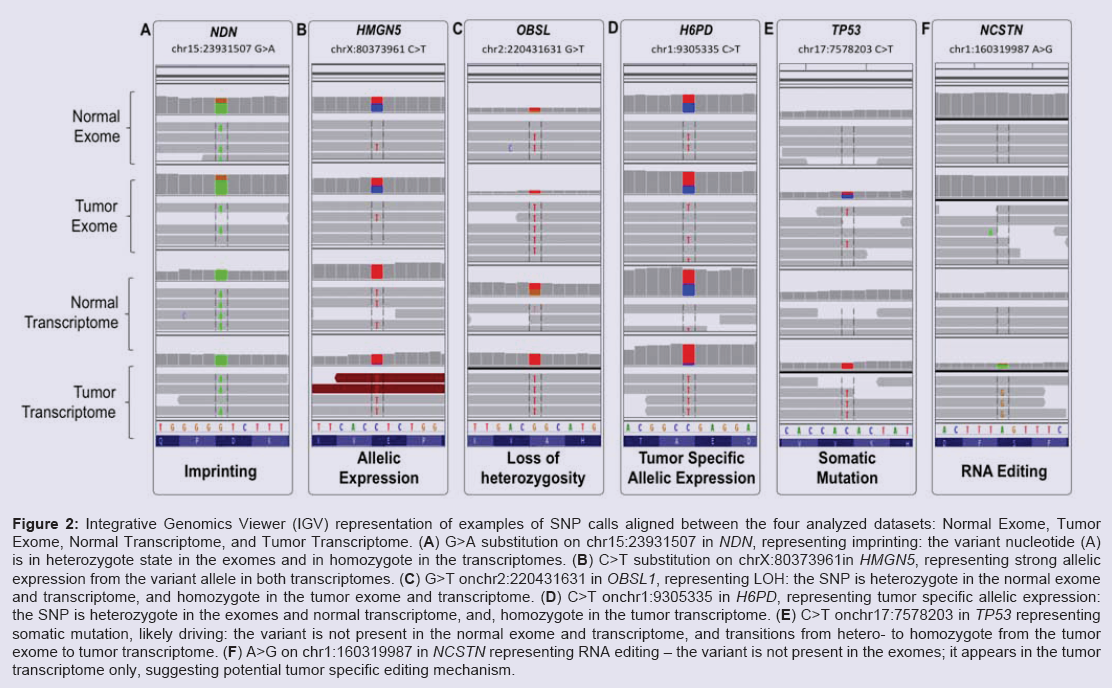
Figure 2: Integrative Genomics Viewer (IGV) representation of examples of SNP calls aligned between the four analyzed datasets: Normal Exome, Tumor Exome, Normal Transcriptome, and Tumor Transcriptome. (A) G>A substitution on chr15:23931507 in NDN, representing imprinting: the variant nucleotide (A) is in heterozygote state in the exomes and in homozygote in the transcriptomes. (B) C>T substitution on chrX:80373961in HMGN5, representing strong allelic expression from the variant allele in both transcriptomes. (C) G>T onchr2:220431631 in OBSL1, representing LOH: the SNP is heterozygote in the normal exome and transcriptome, and homozygote in the tumor exome and transcriptome. (D) C>T onchr1:9305335 in H6PD, representing tumor specific allelic expression: the SNP is heterozygote in the exomes and normal transcriptome, and, homozygote in the tumor transcriptome. (E) C>T onchr17:7578203 in TP53 representing somatic mutation, likely driving: the variant is not present in the normal exome and transcriptome, and transitions from hetero- to homozygote from the tumor exome to tumor transcriptome. (F) A>G on chr1:160319987 in NCSTN representing RNA editing - the variant is not present in the exomes; it appears in the tumor transcriptome only, suggesting potential tumor specific editing mechanism.
Another tumor related SNP preferential expression is defined by variants providing growth advantage to the tumor cells. Such SNPs could be distinguished through comparison between the normal and the tumor transcriptome - while in the normal tissue they are expected to retain their heterozygosity, accelerated growth of SNP-expressing tumor cells will acquire higher proportion of the variant over the wild type alleles. In our dataset, 265 SNPs, six of which not listed in the population databases, satisfied the criteria to be present in heterozygote state in the exomes and the normal transcriptome, vs homozygous expression in the tumor transcriptome. One interesting example is the very rare silent substitution C>T at chr1:9305335(rs138024142, Figure 2D) in the coding sequence of the recently reported to be up-regulated in breast cancer glutamate dehydrogenase H6PD [20]. The variant allele frequency is below 0.01% in general population; parallel in silico modeling of the local genomic region with the wt and the variant nucleotide through the modeling tool “RNAstructure” [21] showed reasonable probability for alteration of the secondary RNA structure harboring the SNP. Focused wet-lab studies are required to assess if rs138024144 variant is implicated in expression regulation of H6PD, in normal or tumor tissues.
Next, we sought to determine the efficiency of the integrative analysis to outline somatic cancer changes. Compared to the extensively performed searches for somatic mutations through comparison of germ line and tumor DNA, RNA-sequencing called SNPs allow estimation of allelic preferential expression, and, thus, uncovering of potential driving (vs passenger) changes [22]. Similarly to above described analyses, among the SNPs confidently called in the tumor (but not in the normal) exome and transcriptome, a particular subset of interest lies in the variants with higher var/wt ratio in the transcriptome, due to the typically low var/wt ratio in the encoding exome. A total of 6 SNPs, none of them present in the population databases, satisfied our criteria for confident somatic mutations. Of note, all of them presented with var/wt ratio >2 in the tumor transcriptome. Of special interest is the missense substitution V216M in the well-known breast cancer oncogene TP53 [23]; the IGV visualization is presented on Figure 2E. The mutation affects the domain required for interaction with FBXO42 and is predicted to be damaging; examination of the IGV files revealed complete homozygous expression in the tumor exome, compared to only several reads in the normal exome. While not present in the population databases, V216M has been reported as a somatic mutation in multiple tumor samples, including breast invasive carcinoma, and is catalogued in the COSMIC database (http://www. sanger.ac.uk/cosmic) [24]. Similarly, we were able to validate all the rest of the somatic calls from our datasets in the COSMIC Database.
RNA editing
To highlight potential RNA editing events, we identified the variants called in transcriptomes but in none of the exomes, with a threshold of var/wt ratio>0.5 (See Figure 1). After removal of the SNPs present in the Exome Variant Server (http://evs.gs.washington. edu/EVS/) [25], 7427 such variants were shared between the tumor and the normal transcriptomes, and additional 2384 were called in the tumor transcriptome only. When we overlaid our results with the known RNA editing events database DARNED [26], a total 1217 variants overlapped with our datasets. Two intriguing observations attracted our attention in thus selected dataset. First, there was apparent gene-centered clustering of RNA-exclusive events, and second, high proportion of the putative RNA editing sites seemed to be predominant, often to monozygotic level, of the variant (vs wt) nucleotide harboring reads. To examine further these observations, we blasted the surroundings of such SNPs against the entire human genome. We found perfect match for the variant bearing allele, in a different genomic locus. An example is the G>A substitution at position chr13:25671320 residing in the coding sequence of PABPC3. Blasting of the SNP calling reads revealed a match at a region of chromosome 8 encoding the highly homologous gene from the same family: PABPC1. Further investigation of more of these sequences suggested that they likely originate from genomic regions highly homologous to the expressed mRNA transcripts, thus representing partial or entire pseudogene-like elements, or closely related homologous genes from the same family/cluster. This explains why these SNP-like calls often cluster in genes - reads derived from an expressed pseudo-gene or homologous gene would generate similar false-positive calls for every mismatch with the original transcript.
To search for new high-confidence RNA editing events, we focused on exonic variants, not listed in population or disease datasets, and not present in DARNED. To remove potentially biased calls, we visually examined the variation appearance through IGV, and removed variants which did not satisfy the criteria for confident RNA editing due to either presence in the exomes (false negative call at the exome level), lack of sufficient exonic coverage, or, called by parts of short read from the intronic 3’- or 5’-splice site. We also blasted the SNP calling reads against the entire genome and retained only SNPs called by uniquely mapped genes. Among the retained high-confidence RNA editing SNPs, one interesting example is the missense substitution S177G (1:160319987 A>G) in the gene encoding nicastrin NCSTN. Of note, this change was called by very few reads in the normal transcriptome, but over-dominated the position in the tumor transcriptome (Figure 2F). Nicastrin cleaves integral membrane proteins, including Notch receptors and betaamyloid precursor protein, and has been recently identified as a cancer driver gene through genome-wide scan [27,28]. In addition, a major recent study has shown that NSCTR regulates breast cancer stem cell properties and tumor growth both in vivo and in vitro [29]. Thus, S177G in NCSTN is worthy focused investigation for cancer driving potential. S177G has never been reported before, however, another NCSTN variant - 1:160327023 A>G - has been reported in DARNED as subject of RNA editing in cerebellum. Notably, both variants represent the common A>I (functional A>G) change, known as the most common RNA editing subject. Taking into account the suggested NCSTN variants involvement in Alzheimer [30,31], there is an apparent need of further investigation of disease implicated NCSTN editing.
Discussion
Despite the fact that the exome and transcriptome target largely overlapping genomic regions, they contain genuinely distinct information layers. While whole exome capture is designed based on the knowledge on all coding genomic sequences, transcriptome does not employ previous knowledge and captures the collection of expressed genes in the studied sample at the moment of harvesting. As such, any single transcriptome represents a snapshot of the transient cell/tissue condition, and only roughly reflects the sample representative genes’ and isoforms’ profile. In terms of number of called variations, two major sources define the significant deviation between the two datasets - the transcriptome will not cover variations in genes that are not expressed, and the exome design does not include most of the large untranslated (UTR), but expressed regions. Additional factors contributing to the exome/transcriptome diversity might be some randomly included non-targeted areas, differences in the sequencing platform, variation call pipeline and filtering criteria.For the regions covered by the exome and transcriptome, the observed deviations usually indicate important regulatory features. In our set of normal/tumor/transcriptome/exome, cancer related changes could be outlined through comparison between the normal and tumor datasets, and expression-specific elements could be found through exome-to-transcriptome comparison (Figure 3). Transcriptome specific allelic expression is an important factor that reflects advantage driven preferential dominance of certain alleles. At nucleotide resolution, it involves one additional information layer - the potential to highlight causative or contributing to the allelic imbalance nucleotide changes. These changes should be carefully distinguished from variants that randomly co-exist along with the driving allele. While lab validation of such findings is definitely a must, application of similar analyses on large-scale can tremendously decrease the rate of random observations and select a finely narrowed feature-set for further analyses.
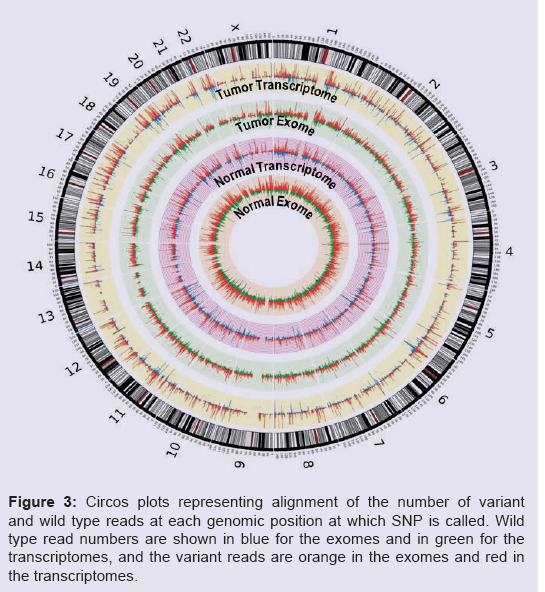
Figure 3: Circos plots representing alignment of the number of variant and wild type reads at each genomic position at which SNP is called. Wild type read numbers are shown in blue for the exomes and in green for the transcriptomes, and the variant reads are orange in the exomes and red in the transcriptomes.
Second, visual examination of the region of interest through IGV or similar genome visualization tool is always helpful to determine the confidence of the call (see Figure 2). Despite the growing number of alignment, assembly and variant calling tools, filtering strategies and confidence-boosting algorithms, false positive and false negative variation calls are still a challenge for NGS.
Third, inherent feature of the short reads sequencing technologies is the possibility for mis-alignment. In our analysis, this bias is illustrated in the RNA-editing focused pipeline. When aligning genomes to transcriptomes, it is essential to keep in mind that similar expressed sequences usually derived from homologous or pseudogenes, can almost perfectly match to the transcript of interest, mistaking for SNP a single mismatch between the transcript of interest and the original site. This is even more emphasized for the non-coding parts of the genome, which still lack sufficient population data and the reference often contains rare variants disfavoring the mapping of the reads to their original site. One approach that definitely restricts such miscalls is local alignment using tools such as BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat?hgsid=340320355&c ommand=start) of the variant calling reads against the whole human genome [32,33]. If a perfect match with another genomic location is found, the SNP call should be treated with increased caution and validated through alternative means.
The purpose of this report is to illustrate approaches that can extract or validate important molecular features, such as expression or growth advantage, imprinting, LOH, somatic changes, RNA editing, through alignment of SNP calls in their allelic context at exome and transcriptome level from the same individual (see Table 3). One additional advantage of the multi-NGS datasets format from the same sample is that it provides means to validate rare or unique findings in cases where no new sample collection is possible. Further, each of the exemplified analytical pipelines can be separately developed to rigorously define the corresponding type of changes in a particular transcriptome dataset of interest. Moreover, the proposed SNP-based pipelines can be integrated with expression information derived from the transcriptome. Interlinking variation and expression require multiple samples, and is outside of the scope of our singleindividual based analysis. In a multi-sample large scale analysis, it holds tremendous potential to uncover regulatory networks through analysis for co-existing and mutually exclusive features.
Acknowledgments
This work is supported by Award Number UL1TR000075 from the NIH National Center for Advancing Translational Sciences and the Clinical and Translational Science Institute at Children’s National Medical Center (CTSI-CN) to Anelia Horvath. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. We also wish to thank the TCGA investigators who generated the data and the bioinformatics group that made this data available. This study adheres to TCGA data usage and publications guidelines (http://cancergenome.nih.gov/abouttcga/policies/policiesguidelines).References
- Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, et al. (2013) Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 121: 4541-4550.
- Kim SC, Jung Y, Park J, Cho S, Seo C, et al. (2013) A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS One 8: e55596.
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, et al. (2013) The genetic landscape of high-risk neuroblastoma. Nat Genet 45: 279-284.
- Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, et al. (2012) Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet 44: 1316-1320.
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, et al. (2012) Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44: 1111-1116.
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, et al. (2012) Recurrent R-spondin fusions in colon cancer. Nature 488: 660-664.
- 7. Gelderman G, Contreras LM (2013) Discovery of posttranscriptional regulatory RNAs using next generation sequencing technologies. Methods Mol Biol 985: 269-295.
- Livyatan I, Harikumar A, Nissim-Rafinia M, Duttagupta R, Gingeras TR, et al. (2013) Non-polyadenylated transcription in embryonic stem cells reveals novel non-coding RNA related to pluripotency and differentiation. Nucleic Acids Res 41: 6300-6315.
- Moore LM, Kivinen V, Liu Y, Annala M, Cogdell D, et al. (2013) Transcriptome and small RNA deep sequencing reveals deregulation of miRNA biogenesis in human glioma. J Pathol 229: 449-459.
- Busche S, Ge B, Vidal R, Spinella JF, Saillour V, et al. (2013) Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res 73: 4323-4336.
- Lasalle JM (2013) Epigenomic strategies at the interface of genetic and environmental risk factors for autism. J Hum Genet 58: 396-401.
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357-359.
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105-1111.
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511-515.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078-2079.
- Horvath A, Pakala SB, Mudvari P, Reddy SD, Ohshiro K, et al. (2013) Novel Insights into Breast Cancer Genetic Variance through RNA Sequencing. Sci Rep 3: 2256.
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178-192.
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, et al. (2011) Integrative genomics viewer. Nat Biotechnol 29: 24-26.
- Hanel ML, Lau JC, Paradis I, Drouin R, Wevrick R (2005) Chromatin modification of the human imprinted NDN (necdin) gene detected by in vivo footprinting. J Cell Biochem 94: 1046-1057.
- Kim S, Kim do H, Jung WH, Koo JS (2013) Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr Relat Cancer 20: 339-348.
- Reuter JS, Mathews DH (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129.
- Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, et al. (2010) Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 107: 18545-18550.
- Walerych D, Napoli M, Collavin L, Del Sal G (2012) The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 33: 2007-2017.
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, et al. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945-950.
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64-69.
- Kiran A, Baranov PV (2010) DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26: 1772-1776.
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, et al. (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407: 48-54.
- Woo HG, Park ES, Lee JS, Lee YH, Ishikawa T, et al. (2009) Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer Res 69: 4059-4066.
- Lombardo Y, Filipovic A, Molyneux G, Periyasamy M, Giamas G, et al. (2012) Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci U S A 109: 16558-16563.
- Ma Z, Han D, Zuo X, Wang F, Jia J (2009) Association between promoter polymorphisms of the nicastrin gene and sporadic Alzheimer's disease in North Chinese Han population. Neurosci Lett 458: 136-139.
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39: 17-23.
- Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, et al. (2012) Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30: 253-260.
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, et al. (2013) Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128-132.