
Journal of Clinical & Medical Case Reports
Early-Infantile Galactosialidosis: Clinical and Radiological Findings
Callyn B. Riggs1, Luis F. Escobar2*, and Megan E. Tucker2
- 1Department of Pediatrics, Peyton Manning Children’s Hospital, Indianapolis, IN 46260, USA
- 2Medical Genetics & Neurodevelopmental Center, Peyton Manning Children’s Hospital, Indianapolis, IN 46260, USA
*Address for Correspondence: Luis F. Escobar, MD, Medical Genetics & Neurodevelopmental Center, Peyton Manning Children’s Hospital, 8402 Harcourt Rd, #300, Indianapolis, IN 46260, USA, Tel: 317-338-5288; Fax: 317-338-7154; E-mail: LFEscoba@stvincent.org
Citation: Riggs CB, Escobar LF, Tucker ME. Early-Infantile Galactosialidosis: Clinical and Radiological Findings. J Clin Med Case Reports. 2015;2(2): 4.
Copyright © 2015 Escobar et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 2
Submission: 19 August 2015 | Accepted: 29 October 2015 | Published: 03 November 2015
Abstract
Galactosialidosis is a rare lysosomal storage disorder. Very few reports of the early-infantile (EIGS) form can be found in the literature. This form typically presents with prenatal non-immune fetal hydrops associated with various complex postnatal clinical findings. We discuss here a case of EIGS seen in our center due to prenatal diagnosis of non-immune hydrops with significant abdominal ascites. Postnatally, the patient was found to have persistent ascites, multiple congenital anomalies, cholestasis, and thrombocytopenia. Radiologic findings suggested osteochondrodysplasia, which in association with the other clinical findings indicated the possibility of a lysosomal storage disorder, such as early-infantile galactosialidosis. We review and discuss here the clinical and radiographic findings in EIGS as described in the literature and compare them to our case study. The information presented should raise awareness of the unusual presentation of early-infantile galactosialidosis and aid neonatologists in the early recognition of this rare and devastating disorder.Keywords
Galactosialidosis; Non-immune hydrops; Dysostosis; Neonatal ascitesIntroduction
Galactosialidosis is an autosomal recessive lysosomal storage disease associated with a combined deficiency of enzymes neuraminidase 1 (NEU1) and β-galactosidase (β-GAL). The prevalence of galactosialidosis is unknown; more than 100 cases have been reported with the majority consisting of the juvenile and adult forms. The early infantile form is the most severe and has been reported in 35 cases. These patients have been diagnosed between birth and three months of age with typical findings of non-immune fetal hydrops, coarse facies, neonatal edema, proteinuria, inguinal hernias and telangiectasias [1]. Hydrops fetalis is typically identifiedon prenatal ultrasound, along with signs of fetal edema and visceromegaly. The skeletal abnormalities previously documented in EIGS are dysostosis multiplex, stippled epiphyses, osteoporosis with coarse trabeculation and significant periosteal reactions [2].Case Report
We report a newborn patient born to a 23 year old G3P0 mother and a 26 year old father, both of Indian (Gujurat) descent at 30 weeks plus 5 days gestational age. There was a history of maternal beta-thalassemia trait, with negative hemoglobin electrophoresis testing for the father. Both parents were otherwise healthy but subsequently found to be likely consanguineous after significant loss of heterozygosity across chromosomes was identified by SNP array. Family history was negative for birth defects, mental retardation, or known chromosomal disorders. The mother had two previous spontaneous abortions. Pregnancy was complicated by nonimmune hydrops fetalis, and prenatal ultrasound evidence of echogenic bowel, ventriculomegaly, short bones, significant abdominal ascites, and a 2-vessel cord. In order to facilitate delivery fetal peritoneal aspiration was performed immediately prior to cesarean section delivery which also help to decrease diaphragmatic and pulmonary compression and allowed improve pulmonary function. Close to 150 ml of amber colored fluid was removed. The infant was delivered emergently via cesarean section due to breech presentation and variable fetal heart rate decelerations with reversed diastolic flow. Apgar scores were 2 at 1 minute, 5 at 5 minutes, and 8 at 10 minutes. Meconium stained fluid was present at delivery. The infant appeared cyanotic with respiratory depression, underwent tracheal suctioning then endotracheal intubation with surfactant administration at 10 minutes of life. He did not require chest compressions during resuscitation. Chest radiograph shortly after birth revealed diminished lung volumes with diffuse granular opacities, thus infant was admitted to the Neonatal Intensive Care Unit and placed on high-frequency oscillatory ventilation (HFOV).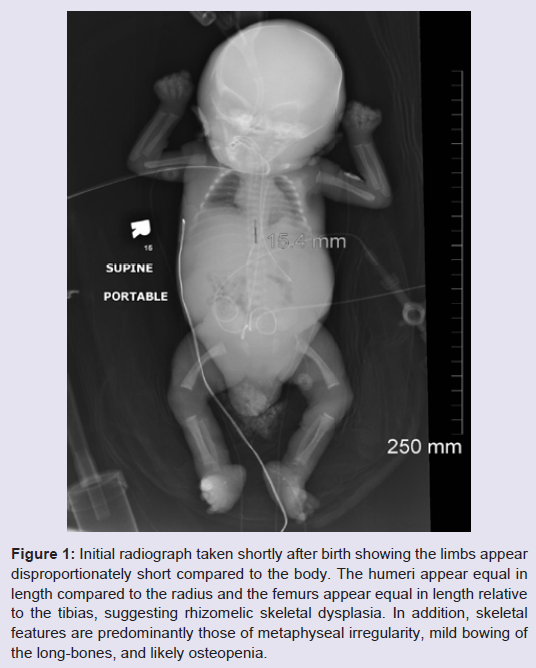
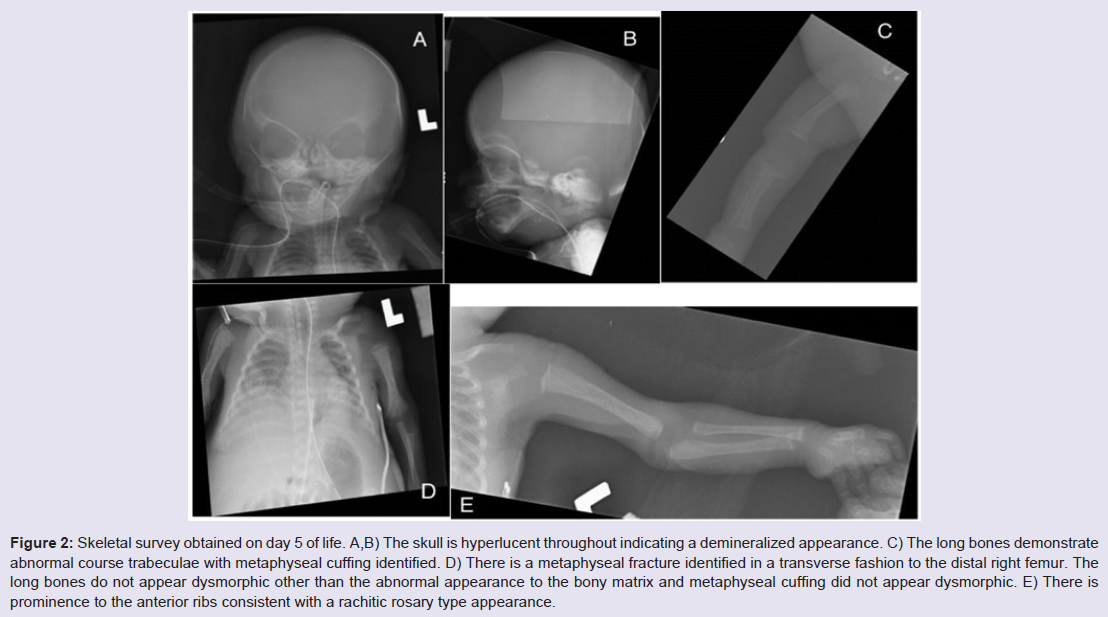
Additional lab abnormalities found in this case were persistent neutropenia with initial absolute neutrophil count of 605 and remaining below 1000 for his entire course, in addition to anemia and thrombocytopenia requiring multiple blood and platelet transfusions. Urine testing for cytomegalovirus was negative. The patient’s hospital course continued to be complicated by respiratory distress with several failed attempts at extubation. He remained on antibiotics since birth due to concerns for infection in the setting of pancytopenia. Significant abdominal ascites persisted with abdominal girth increasing from 23 to 32 cm. Peritoneal aspirations were not performed due to the expectation that fluid would re-accumulate and an aspiration would only provide temporary relief. The decision was made to withdraw life support and the patient died at 38 days of life.
Discussion
The clinical findings of this case, particularly non-immune hydrops fetalis, suggest the diagnosis of a storage disorder, however it can be difficult to differentiate between the numerous lysosomal storage disorders based on phenotypic findings alone. Other genetic conditions causing non-immune fetal hydrops include Gaucher’s disease, generalized gangliosidosis, Salla’s disease, sialidosis, mucopolysaccharidosis types IV and VII, and Tay-Sach’s disease. Galactosialidosis shares many of these physical findings with other lysosomal storage disorders, such as coarse facies, edema, abnormal bone formation and cherry red spots on eye exam [4]. Visceromegaly is another common finding of storage disorders, however hepatosplenomegaly is more commonly seen in EIGS rather than enlargement of the heart or kidneys. There have been 14 cases of EIGS that report heart involvement, typically cardiomegaly, thickened septum and subsequent cardiac failure [1]. Kidney involvement usually manifests at proteinuria, and kidney enlargement has not been documented in cases of galactosialidosis, but has been reported in cases of glycogen storage disease type I and muchopolysaccharidoses [5].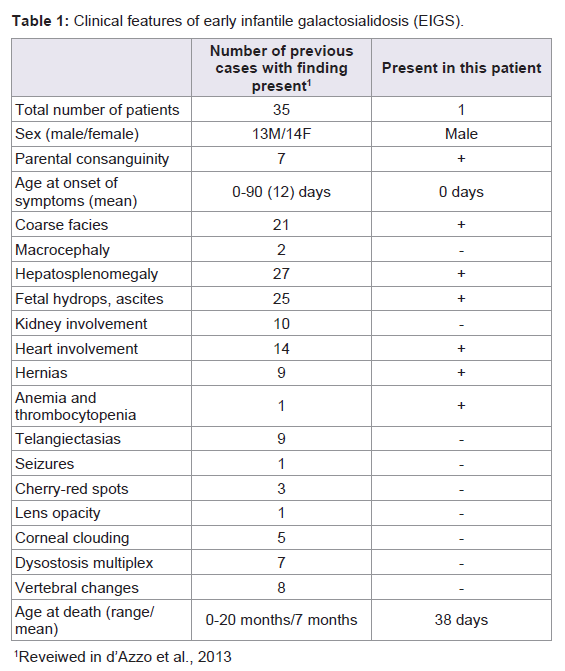
References
- d'Azzo A, Andria G, Strisciuglio P, Galjaard H (2001) Galactosialidosis. In, Scriver CR, Beaudet AL, Sly WS, Valle D (eds.). The metabolic & molecular bases of inherited disease. Vol. III (8th edn) New York: McGraw-Hill, pp: 3811.
- Patel MS, Callahan JW, Zhang S, Chan AK, Unger S, et al. (1999) Early-infantile galactosialidosis: prenatal presentation and postnatal follow-up. Am J Med Genet 85: 38-47.
- Caciotti A, Catarzi S, Tonin R, Lugli L, Perez CR, et al. (2013) Galactosialidosis: review and analysis of CTSA gene mutations. Orphanet J Rare Dis 8: 114.
- Carvalho S, Martins M, Fortuna A, Ramos U, Ramos C, et al. (2009) Galactosialidosis presenting as nonimmue fetal hydrops: a case report. Prenat Diagn 29: 895-896.
- Schwartz IV, Artigalas O, Ries M, Clarke JT, Giugliani R, et al. (2009) Punctate calcifications in lysosomal storage disorders. Clin Dysmorphol 18: 172-177.
- Offiah AC, Hall CM (2003) Radiological diagnosis of the constitutional disorders of bone. As easy as A, B, C? Pediatr Radiol 33: 153-161.
- Sewell AC, Pontz BF, Weitzel D, Humburg C (1987) Clinical heterogeneity in infantile galactosialidosis. Eur J Pediatr 146: 528-531.