
Journal of Cytology & Molecular Biology
Download PDF
Research Article
*Address for Correspondence: Maqsood A. Chotani, Center for Cardiovascular & Pulmonary Research, The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, USA, E-mail: Maqsood.Chotani@gmail.com
Citation: Motawea HK, Blazek AD, Zirwas MJ, Pleister AP, Ahmed AAE, et al. Delocalization of Endogenous A-kinase Antagonizes Rap1-Rho-α2C-Adrenoceptor Signaling in Human Microvascular Smooth Muscle Cells. J Cytol Molecul Biol. 2014;1(1): 9.
Copyright © 2014 Motawea HK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Cytology & Molecular Biology | ISSN: 2325-4653 | Volume: 1, Issue: 1
Submission: 07 November 2013 | Accepted: 06 January 2014 | Published: 10 January 2014
Reviewed & Approved by: Dr. Annie Angers, Department of Sciences, University of Montreal, Canada
 Arterioles play a vital role in modulating blood flow to tissues and organs and generate peripheral vascular resistance [7]. Arterioles have high wall-to-lumen ratio compared to other vessels, with majority of the components of the wall being the circumferential smooth muscle cells. The smooth muscle layer allows the arteriole to vary wall tension and diameter, and consequently resistance of the vessel. These alterations in resistance contribute to perfusion of the vascular beds. Further, vascular stress or injury may trigger an inflammatory response leading to increased release of prostaglandin E2 (PGE2) and prostacyclin (PGI2), activation of Gs-coupled receptors, and increased intracellular cAMP. Understanding the role of cAMP in these arterioles therefore, has relevance to vessel lumen diameter and blood flow control in physiology and pathophysiology.
Arterioles play a vital role in modulating blood flow to tissues and organs and generate peripheral vascular resistance [7]. Arterioles have high wall-to-lumen ratio compared to other vessels, with majority of the components of the wall being the circumferential smooth muscle cells. The smooth muscle layer allows the arteriole to vary wall tension and diameter, and consequently resistance of the vessel. These alterations in resistance contribute to perfusion of the vascular beds. Further, vascular stress or injury may trigger an inflammatory response leading to increased release of prostaglandin E2 (PGE2) and prostacyclin (PGI2), activation of Gs-coupled receptors, and increased intracellular cAMP. Understanding the role of cAMP in these arterioles therefore, has relevance to vessel lumen diameter and blood flow control in physiology and pathophysiology.
Delocalization of Endogenous A-kinase Antagonizes Rap1-Rho-α2C-Adrenoceptor Signaling in Human Microvascular Smooth Muscle Cells
Hanaa K. B. Motawea1,2, Alisa D. Blazek3, Matthew J. Zirwas4, Adam P. Pleister4, Amany A. E. Ahmed2, Bradley K. McConnell5 and Maqsood A. Chotani1,3,6*
- 1Center for Cardiovascular & Pulmonary Research, The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, USA
- 2Faculty of Pharmacy, Department of Pharmacology & Toxicology, Helwan University, Helwan, Egypt
- 3Davis Heart and Lung Research Institute, Ohio State University, Columbus, Ohio, USA
- 4Internal Medicine, Ohio State University Wexner Medical Center, Columbus, Ohio, USA
- 5Department of Pharmacological and Pharmaceutical Sciences, University of Houston, Texas Medical Center Houston, Texas, USA
- 6Department of Pediatrics, Ohio State University, Columbus, Ohio, USA
*Address for Correspondence: Maqsood A. Chotani, Center for Cardiovascular & Pulmonary Research, The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio, USA, E-mail: Maqsood.Chotani@gmail.com
Citation: Motawea HK, Blazek AD, Zirwas MJ, Pleister AP, Ahmed AAE, et al. Delocalization of Endogenous A-kinase Antagonizes Rap1-Rho-α2C-Adrenoceptor Signaling in Human Microvascular Smooth Muscle Cells. J Cytol Molecul Biol. 2014;1(1): 9.
Copyright © 2014 Motawea HK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Cytology & Molecular Biology | ISSN: 2325-4653 | Volume: 1, Issue: 1
Submission: 07 November 2013 | Accepted: 06 January 2014 | Published: 10 January 2014
Reviewed & Approved by: Dr. Annie Angers, Department of Sciences, University of Montreal, Canada
Abstract
The second messenger cyclic AMP (cAMP) plays a vital role in the physiology of the cardiovascular system, including vasodilation of large blood vessels. This study focused on cAMP signaling in peripheral blood vessels, specifically in human vascular smooth muscle (microVSM) cells explanted from skin punch biopsy arterioles (also known as resistance vessels) of healthy volunteers. Using these human microVSM we recently demonstrated cAMP activation of exchange protein activated by cAMP (Epac), the Ras-related small GTPase Rap1A, and RhoA-ROCK-F-actin signaling in human microVSM to increase expression and cell surface translocation of functional α2C-adrenoceptors (α2C-ARs) that mediate vasoconstriction. Protein-protein association with the actin-binding protein filamin-2 and phosphorylation of filamin-2 Ser2113 by cAMP-Rap1A-Rho-ROCK signaling were necessary for receptor translocation in these cells. Although cAMP activated A-kinase in these cells, these effects were independent of A-kinase, and suggested compartmentalized A-kinase local signaling facilitated by A-kinase anchoring proteins (AKAPs). In this study we globally disrupted A-kinase-AKAP interactions by the anchoring inhibitor decoy peptide Ht31 and examined the effect on α2C-AR expression, translocation, and function in quiescent microVSM treated with the adenylyl cyclase activator and cAMP elevating agent forskolin. The results show that Ht31, but not the control peptide Ht31-P, reduced forskolin-stimulated Ser133 phosphorylation of A-kinase substrate CREB, reduced α2C-AR mRNA levels, reduced cell surface translocated receptors, and attenuated agonist-triggered receptor functional responses. Together, the results suggest that compartmentalized cAMP signaling elicits a selective cellular response in microVSM, which may have relevance to arteriole physiological function and responses.Keywords
Microcirculation; Arteriole; Resistance vessel; Vasoconstriction; Vascular smooth muscle; Signal transduction; cAMP; Ht31; UK 14,304Abbreviations
AC: Adenylyl Cyclase; A-kinase: Protein Kinase A; AKAP: A-Kinase Anchoring Protein; ATP: Adenosine Triphosphate; α2C-ARs: α2C-Adrenoceptors; cAMP: 3′, 5′ Cyclic Adenosine Monophosphate; CREB: Cyclic AMP Response Element Binding Protein; Epac: Exchange Protein Activated by Cyclic AMP; FSK: Forskolin; GAPDH: Glyceraldehyde 3-Phosphate Dehydrogenase; GTP: Guanosine 5′-Triphosphate; microVSM: Microvascular Smooth Muscle Cells; RNase protection assays: Ribonuclease Protection Assays; UK 14,304: 5-Bromo-N-(2-Imidazolin-2-Yl)-6-Quinoxalinamine; IBMX: 3-Isobutyl-1-MethylxanthineIntroduction
The second messenger 3′, 5′ cyclic adenosine monophosphate (cAMP) plays a vital role in intracellular signaling in the cardiovascular system [1]. This molecule is synthesized by activated adenylyl cyclase from adenosine triphosphate (ATP) in response to extracellular stimulation by G-stimulatory (Gs)-coupled receptors. In smooth muscle cells of the vasculature, increased intracellular levels of cAMP lead to vasodilation of large blood vessels, including aorta and pulmonary artery [2]. However, in smooth muscle cells derived from smaller vessels such as arterioles (microvascular smooth muscle cells or microVSM), cAMP has a unique role in increasing expression and cell surface translocation of α2C-adrenoceptors (α2C-ARs), known to mediate vasoconstriction [3]. In microVSM cAMP activates both A-kinase and the Ras-related small G protein Rap1, and signaling associated with these molecules [3-5].Our studies on microVSM have revealed a unique and crucial role of cAMP in increasing gene expression of the G protein coupled α2C-ARs, activating the RhoA-ROCK pathway and increasing reorganization of filamentous (F) actin. The α2C-ARs are intracellular receptors that reside in a silent state in the perinuclear Golgi region, and upon stimulation with cAMP elevating agents, that mimic vascular stress or injury responses, translocate to the cell surface, where these receptors are functional and elicit the biological response of vasoconstriction [3]. Although cAMP activates both the A-kinase and the Epac-Rap1 pathways in these cells, these effects of cAMP are A-kinase independent and are predominantly mediated by Rap1 subtype A (Rap1A) [3,4]. Indeed, in Rap1A-null microVSM these effects of cAMP are lost, and can be rescued by introducing constitutively activated Rap1A (Rap1A-CA) in the Rap1A-null background. Rap1A-CA increased expression of α2C-ARs and activated the RhoA-ROCK pathway, increasing F-actin [3]. In transiently transfected human microVSM Rap1A-CA transcriptionally increased expression of luciferase-driven α2C-AR promoter (-1915/+5, relative to the transcription start site +1) through JNK-c-Jun signaling [6] and also increased endogenous microVSM receptor levels [3]. The α2C-ARs associate with the actin binding protein filamin-2 via protein-protein interaction. Rap1A-Rho-ROCK signaling phosphorylates filamin-2 Ser2113, which is necessary for receptor translocation to the cell surface [5]. Rap1A therefore, has dual effect on α2C-ARs; it can transcriptionally increase mRNA expression, increasing intracellular levels of the receptor [3,4], and can increase cell surface receptor translocation by activation of RhoA-ROCK-filamin-2 pathway [5], altogether up-regulating receptor function. These studies therefore showed that receptor expression (including transcription, translation) and translocation were separate, independent processes. The cAMP activated serine-threonine A-kinase however, does not show this positive effect in microVSM, as pharmacological inhibition of A-kinase activity by the cell-permeable inhibitor H-89 shows no effect on α2C-AR regulation. The A-kinase however, has the potential to down regulate α2C-ARs. Transient transfection of the A-kinase catalytic subunit decreased basal α2C-AR promoter driven luciferase activity, whereas the cAMP response element (CRE) driven reporter activated by the A-kinase substrate CREB was positively activated [4]. Together, some of the known targets and effects of cAMP on microVSM are summarized in Table 1.
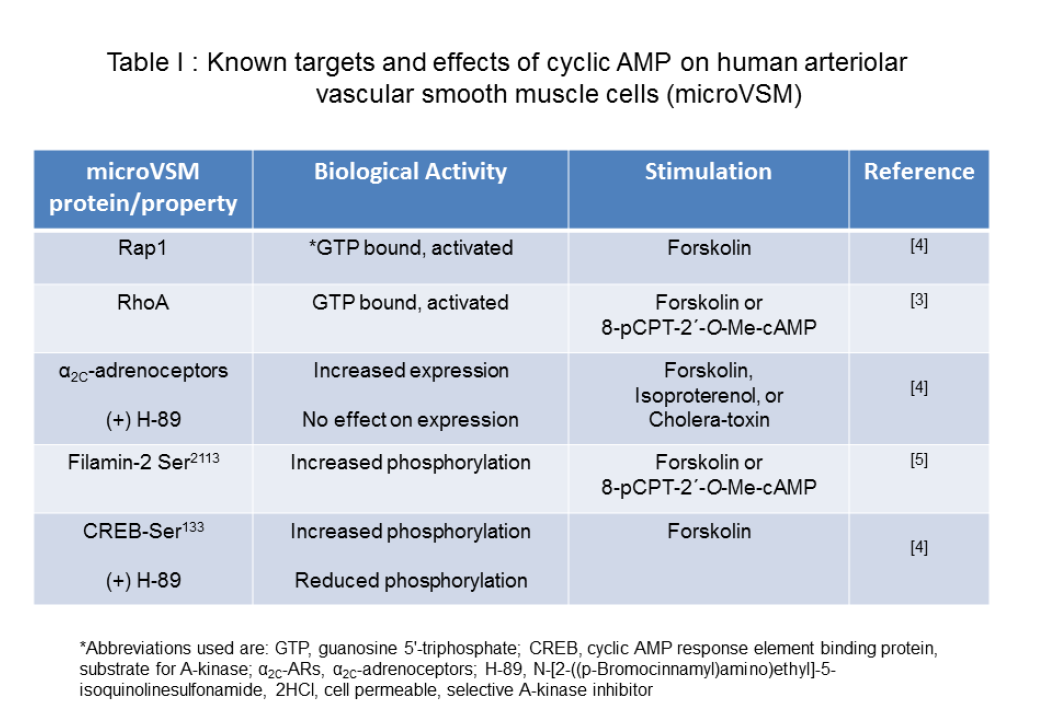
Table 1: Known targets and effects of cyclic AMP on human arteriolar vascular smooth muscle cells (microVSM)
In this study, we utilized the Ht31 decoy peptide to see the impact of delocalized catalytic subunit of A-kinase on microVSM α2C-ARs. Our results suggest that compartmentalized cAMP signaling in microVSM is necessary for resistance vessel physiological function and responses.

Materials and Methods
a. Anti-α2C-adrenoceptor affinity purified rabbit polyclonal antibody was custom generated, and recognizes the ~70 kDa mature receptor form that translocates from the Golgi compartment to the cell surface [8-11].
b. Anti-A-kinase R-II-α subunit (affinity purified rabbit polyclonal) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX).
c. Anti-human filamin (monoclonal, purified by protein A affinity chromatography, clone 1.BB.804) was purchased from US Biological Biochemicals and Biological Reagents (Swampscott, MA).
d. Anti-human and murine filamin-2 phospho-Ser2113specific antibody (rabbit polyclonal, purified by affinity chromatography) was purchased from Invitrogen (Camarillo, CA).
e. Anti-CREB and anti-CREB (pSer133) were detected using the PhosphoPlus® CREB (Ser133) antibody kit (Cell Signaling Technology, Beverly, MA).
f. Anti-HA.11 monoclonal antibody (clone 16B12) was purchased from Covance (Berkeley, CA).
All secondary horseradish peroxidase or alkaline phosphatase labeled (anti-mouse and anti-rabbit) antibodies were purchased from GE Healthcare/Amersham (United Kingdom) or Jackson Immuno Research Laboratories, Inc. (West Grove, PA).
Chemicals:
a. Forskolin: This adenylyl cyclase activator, derived from Coleus forskohlii and ≥98% pure, was obtained in powder form from Sigma (St. Louis, MO). Working stock solutions of 10 mM were made by dissolving in DMSO solvent.
b. UK 14, 304: This synthetic α2-AR agonist was obtained in powder form from Sigma. Working stock solutions of 5 mM were made in DMSO solvent.
c. IBMX: this phosphodiesterase inhibitor was obtained in powder form from Sigma. Working stock solutions of 0.675 M were made in DMSO solvent.
MicroVSM cultures
Human VSM culture: Human arteriolar vascular smooth muscle cells were used for the studies and were explanted from dermal arterioles of healthy volunteers as previously described [10]. The procedures and studies performed on human tissue were approved by the Biomedical Sciences Institutional Review Board (IRB) of the Ohio State University.
Immunofluorescence-confocal microscopy and quantitation of fluorescence
This technique was performed as previously described [3,5]. Briefly, following treatments live cells were fixed in 3% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with primary antibody after blocking with 2% normal goat serum. Alexa Fluorlabeled secondary antibody (Molecular Probes, Life Technologies, Grand Island, NY) was utilized for visualization of signal, and Hoechst stain for visualizing nuclei. Images were captured by random selection in multiple fields using laser scanning confocal microscopy (LSM 510, Zeiss, Germany). Cells that were clearly damaged during the processing and mounting steps were excluded. This approach provides optical slices of microVSM and allows spatial visualization of intracellular and cell boundary associated endogenous α2C-ARs. When comparing fluorescence, the highest intensity observed was used as reference for each set of experiments, and identical settings and conditions were used to capture and process all images. When assessing changes in mean fluorescence intensity, the region of interest tool (NIS-Elements AR™ Laboratory Image Analysis System, Nikon Instruments, Melville, NY) was used for quantitation for each set of experiments. The quantitation of mean fluorescence intensity of cell boundary associated α2C-ARs was assessed at four different regions (chosen at random) of the cell boundary per cell. Previous studies have established that cell boundary associated receptors are cell-surface α2C-ARs [5]. This approach therefore allows relative quantitative assessment of receptor trafficking to the cell surface.
Relative F-actin levels in microVSM were assessed by utilizing fluorescently labeled phalloidin (Molecular Probes, Alexa Fluor®-488 phalloidin). Nuclei were visualized with Hoechst stain. When comparing fluorescence in unstimulated and stimulated cells, the highest intensity was used as reference and all images were captured and processed with identical settings and conditions. All images were processed and presented in their original state, without editing or modifications. Changes in mean fluorescence intensity were quantitated by using the region-of-interest tool, NIS-Elements AR™ Laboratory Image Analysis System (Nikon Instruments Inc.).
Adenovirus constructs Ht-31 and Ht-31-P
Recombinant adenovirus encoding HA-tagged Ht31 peptide (Ad-HA-Ht31418–718; Ht31) or HA-tagged Ht31P (proline-substituted derivative) (Ad-HA-Ht31P418–718; Ht31-P) were generated as previously described [12]. The A-kinase R subunits are anchored by protein-protein interactions to A-kinase anchoring proteins (AKAPs), conferring distinct intracellular localization and specificity of signaling. The AKAPs contain a conserved amphipathic helix of 23 residues (amino acids 493-515) that binds the RII subunit [13-15]. In in vitro and in vivo studies, expression of a peptide containing these conserved residues (referred to as anchoring inhibitor Ht31) leads to competition with R subunit binding or endogenous A-kinase holoenzyme binding, globally delocalizing A-kinase from all AKAPs and decreasing local availability of the catalytic (C) subunit which is associated with the R subunit [12,13,16-19]. Indeed, cAMP can induce catalytic activity of A-kinase without R-C subunit dissociation [20]. The proline substituted peptide (Ht31-P) has two proline substitutions of hydrophobic residues (amino acids 499 (Ala) and 507 (Ile)). These prolines disrupt the secondary structure of the amphipathic helix necessary for RII subunit binding [12]. The expression of Ht31-P therefore, does not block A-kinase/AKAP interaction and serves as a negative control. MicroVSM viraltransductions were optimized using equivalent numbers of virus particles per cell (of each peptide; multiplicity of infection, MOI) for transduction as previously described [3,10]. Peptide expression and stability were confirmed by Western blot analysis using anti-HA antibody directed against the peptide amino terminus tag (~88 hours after transduction). Although both peptides were overexpressed, the effects observed were specific to Ht31, as in control studies increasing MOI of transduction of Ht31-P compared with Ht31 showed no effect on A-kinase delocalization. These observations were consistent with previous studies that confirmed and established in vivo peptide stability and specificity [12]. All virus work was performed according to NIH Guidelines and Institutional Biological & Chemical Safety Committee (IBCSC) guidelines and approved protocols.
Quantitative ribonuclease (RNase) protection assays (qRPA)
This assay was developed to quantitate α2-AR subtype expression in human tissue and explanted microVSM. The reliability and specificity of this assay has therefore been established in previous studies [4,10]. These assays were performed as previously described using an internal control for the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [4,10,21]. The α2C-AR was detected as a 348 nucleotide protected fragment, and the GAPDH as a 433 nucleotide protected fragment. The data were normalized to GAPDH.
Western blotting
This was performed as previously described [3]. Samples were separated on 10% pre-cast gels (Criterion™ Tris-HCl, Bio-Rad Laboratories, Hercules, CA) and immunoblotted for P-CREB and CREB. The results for P-CREB were normalized to total CREB expression, which was used as control for sample loadings. The images were scanned for quantitation using Quantity One software (Bio-Rad Laboratories).
Measurement of intracellular cAMP
The intracellular cyclic AMP levels in human microVSM transduced with Ht31-P or Ht31 were measured to assess endogenous α2C-AR cell surface association and function. Cyclic AMP levels were quantitated using ELISA (following manufacturer recommendations, Enzo Life Sciences Inc. Farmingdale, NY). The assays were performed as previously described [3].
Statistical analysis
Statistical analysis of the data was performed by Student’s t-test for either paired or unpaired observations. When more than two means were compared, either a one-way ANOVA with Bonferroni or Dunnett’s post hoc test for multiple comparisons or a two-way ANOVA with Tukey-Kramer’s post hoc test (Graph Pad Prism, San Diego, CA) was used to identify differences among groups. Data are presented as means ± SE, where n equals the number of independent experiments or individual cells. Values were considered statistically different when P≤ 0.05.

 Ht31 and effect on α2C-adrenoceptor expression, translocation, and function
Ht31 and effect on α2C-adrenoceptor expression, translocation, and function
 Previous studies also established the feasibility of utilizing immunofluorescence-confocal imaging to examine spatial and temporal distribution of endogenous microVSM α2C-ARs, actin filaments, and phospho-Ser2113 filamin-2 in quiescent or stimulated cells [3,5]. Studies showed that α2C-ARs co-localized with actin filaments and the cell boundary in stimulated cells. Punctate staining of α2C-ARs (shown to be in intracellular vesicular compartments) was observed along actin filaments indicating vesicular transport of α2C- ARs to the cell boundary. Utilizing amino-terminus tagged α2C-ARs, these cell boundary receptors were determined to be associated with the cell surface and were functional [3,5]. The anti-α2C-adrenoceptor antibody used in the studies recognizes the ~70 kDa mature receptor form that translocates from the Golgi compartment to the cell surface [8-11]. Further, filamin-2 was shown to be necessary for receptor translocation to the cell surface; phosphorylated Ser2113 filamin-2 was associated with filaments and cell boundary in stimulated cells [5].
Previous studies also established the feasibility of utilizing immunofluorescence-confocal imaging to examine spatial and temporal distribution of endogenous microVSM α2C-ARs, actin filaments, and phospho-Ser2113 filamin-2 in quiescent or stimulated cells [3,5]. Studies showed that α2C-ARs co-localized with actin filaments and the cell boundary in stimulated cells. Punctate staining of α2C-ARs (shown to be in intracellular vesicular compartments) was observed along actin filaments indicating vesicular transport of α2C- ARs to the cell boundary. Utilizing amino-terminus tagged α2C-ARs, these cell boundary receptors were determined to be associated with the cell surface and were functional [3,5]. The anti-α2C-adrenoceptor antibody used in the studies recognizes the ~70 kDa mature receptor form that translocates from the Golgi compartment to the cell surface [8-11]. Further, filamin-2 was shown to be necessary for receptor translocation to the cell surface; phosphorylated Ser2113 filamin-2 was associated with filaments and cell boundary in stimulated cells [5].
 The spatial distribution and quantitation of changes in mean fluorescence intensity (as a measure of changes in phosphorylation status) of phospho-Ser2113 filamin-2 showed a significant increase at 9 h of forskolin stimulation compared with 0 h, in both Ht31-P and Ht31 transduced cells. The phospho-Ser2113 filamin-2 was associated with filaments and cell boundaries in stimulated cells. There were no significant differences observed at 9 h for Ht31-P versus Ht31. However, a significant difference was observed at 16 h of forskolin stimulation in Ht31 transduced cells versus Ht31-P (Figure 4G and Figure 4H). Finally, the impact of Ht31 on receptor biological function was assessed. The α2C-ARs are Gi-coupled receptors and upon activation by agonist cell surface receptors inhibit intracellular cAMP produced by adenylyl cyclase [29]. With increasing agonist concentration modest to strong biological responses were observed in cells transduced with Ht31-P, consistent with increased cell surface-associated receptors (Figure 5A). However, with increasing agonist concentration, no responses or attenuated biological responses were observed in cells transduced with Ht31, consistent with reduced cell surface-associated receptors (Figure 5B).
The spatial distribution and quantitation of changes in mean fluorescence intensity (as a measure of changes in phosphorylation status) of phospho-Ser2113 filamin-2 showed a significant increase at 9 h of forskolin stimulation compared with 0 h, in both Ht31-P and Ht31 transduced cells. The phospho-Ser2113 filamin-2 was associated with filaments and cell boundaries in stimulated cells. There were no significant differences observed at 9 h for Ht31-P versus Ht31. However, a significant difference was observed at 16 h of forskolin stimulation in Ht31 transduced cells versus Ht31-P (Figure 4G and Figure 4H). Finally, the impact of Ht31 on receptor biological function was assessed. The α2C-ARs are Gi-coupled receptors and upon activation by agonist cell surface receptors inhibit intracellular cAMP produced by adenylyl cyclase [29]. With increasing agonist concentration modest to strong biological responses were observed in cells transduced with Ht31-P, consistent with increased cell surface-associated receptors (Figure 5A). However, with increasing agonist concentration, no responses or attenuated biological responses were observed in cells transduced with Ht31, consistent with reduced cell surface-associated receptors (Figure 5B).
Results
Delocalization of endogenous A-kinase using anchoring inhibitor decoy peptide Ht31The A-kinase holoenzyme, comprising two catalytic (C) subunits held inactive by association with two regulatory (R) subunits, mediates cAMP signaling [22]. cyclic AMP binding to R subunits uncovers the biological activity of the C subunits, leading to selective phosphorylation of substrate proteins. The R subunits are anchored by protein-protein interactions to A-kinase anchoring proteins (AKAPs), conferring distinct intracellular localization and specificity of signaling, important for the maintenance of cellular function [13,23-26]. To examine the role of A-kinase on endogenous α2C-ARs in human microVSM, experiments were conducted using the anchoring inhibitor decoy peptide Ht31 to delocalize endogenous A-kinase and to measure the impact on α2C-AR expression, translocation, and function.
In microVSM, the endogenous RII subunit is sequestered in a juxtanuclear sac-like organelle region, examined with immunofluorescence-confocal microscopy staining, reflecting RIIAKAP distribution in these cells, similar to non-VSM cells [27,28]. Expression of Ht31-P (even with increasing multiplicity of infection compared with Ht31) showed no effect on RII subunit distribution (red, Alexa Fluor-568, Figure 1A). However, expression of Ht31disrupted this localization and showed diffuse staining. The RII staining was present in the cytosol and cell boundary (Figure 1A). Quantitative assessment of RII distance spread relative to the nucleus in Ht31-P or delocalized in Ht31 was measured in DMSO or FSK treated cells. Using this approach significant delocalization of RII was observed in cells with Ht31 versus Ht31-P (Figure 1B). Delocalization will reduce the local availability of the C subunit for substrates including CREB. The effect of delocalization was therefore assessed by examining phosphorylation of the A-kinase substrate CREB at Ser133. CREB is phosphorylated at Ser133 as early as 10 min of stimulation by forskolin [4]. Significantly reduced phosphorylation of CREB Ser133 was noted for cells transduced with Ht31 versus Ht31-P at 10 min, 30 min, and 60 min of forskolin stimulation (Figure 2).
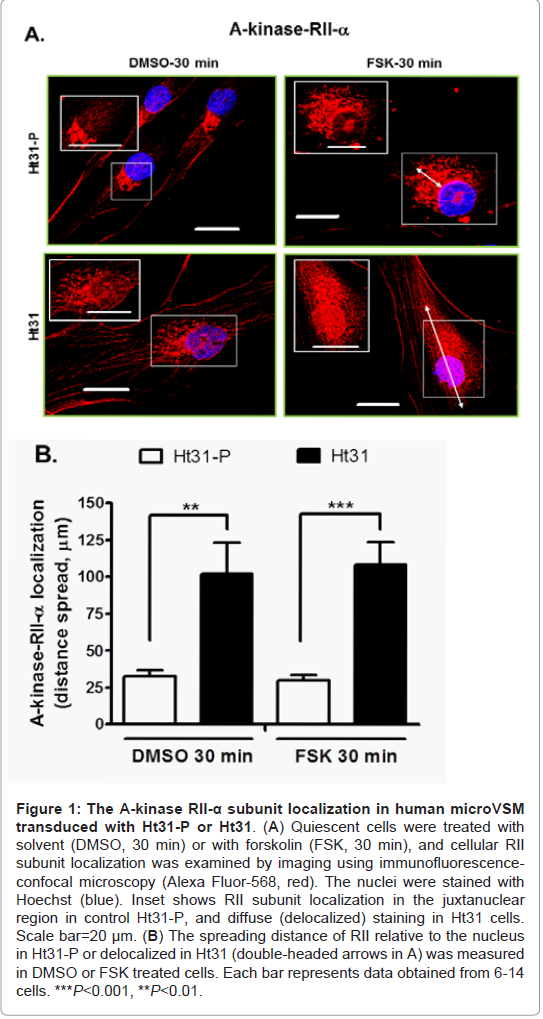
Figure 1: The A-kinase RII-α subunit localization in human microVSM transduced with Ht31-P or Ht31.(A) Quiescent cells were treated with solvent (DMSO, 30 min) or with forskolin (FSK, 30 min), and cellular RII subunit localization was examined by imaging using immunofluorescence confocal microscopy (Alexa Fluor-568, red). The nuclei were stained with Hoechst (blue). Inset shows RII subunit localization in the juxtanuclear region in control Ht31-P, and diffuse (delocalized) staining in Ht31 cells. Scale bar=20 μm. (B) The spreading distance of RII relative to the nucleus in Ht31-P or delocalized in Ht31 (double-headed arrows in A) was measured in DMSO or FSK treated cells. Each bar represents data obtained from 6-14 cells. ***P<0.001, **P<0.01.
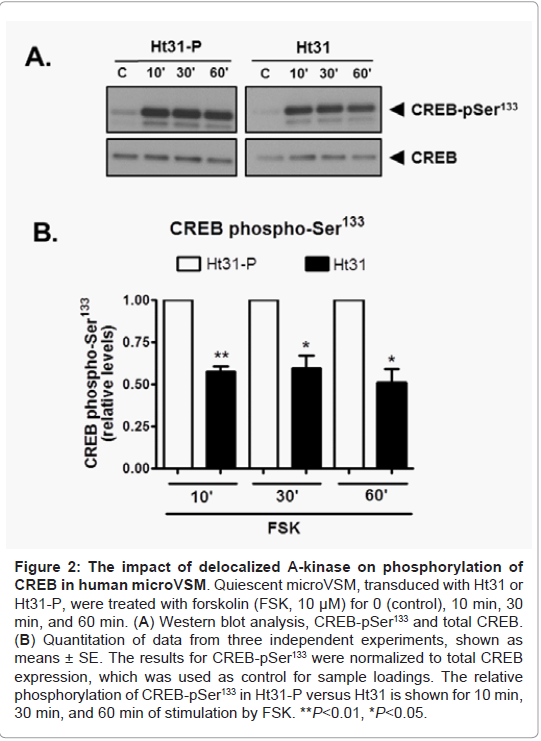
Figure 2: The impact of de-localized A-kinase on phosphorylation of CREB in human microVSM. Quiescent microVSM, transduced with Ht31 or Ht31-P, were treated with forskolin (FSK, 10 μM) for 0 (control), 10 min, 30 in, and 60 min. (A) Western blot analysis, CREB-pSer133 and total CREB. (B) Quantitation of data from three independent experiments, shown as means ± SE. The results for CREB-pSer133 were normalized to total CREB xpression, which was used as control for sample loadings. The relative phosphorylation of CREB-pSer133 in Ht31-P versus Ht31 is shown for 10 min, 30 min, and 60 min of stimulation by FSK. **P<0.01, *P<0.05.
Previous studies established the riboprobe specificity for α2C-AR mRNA in quantitative RNase protection assays [10]. Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control for sample loadings, allowing quantitation and normalization of the data. This approach allowed quantitative assessment of the time course of α2C-AR mRNA induction upon forskolin (10 μM) stimulation. The studies showed that α2C-AR mRNA is induced by forskolin, reaching maximum levels at 6 h of stimulation, and is not affected by the A-kinase pharmacological inhibitor H-89 [4]. This increase in α2C-AR mRNA led to a corresponding increase in the mature glycosylated receptor protein levels, which reached highest detectable levels at 16 h of forskolin stimulation [3,6,10]. In the present study however, expression of Ht31 affected basal (unstimulated) and forskolin (FSK, 10 μM) stimulated induction of α2C-ARs in human microVSM (examined at 6 h of FSK stimulation), compared with expression in the presence of control Ht31-P, which was taken as reference, 100% (Figure 3A, Figure 3B).
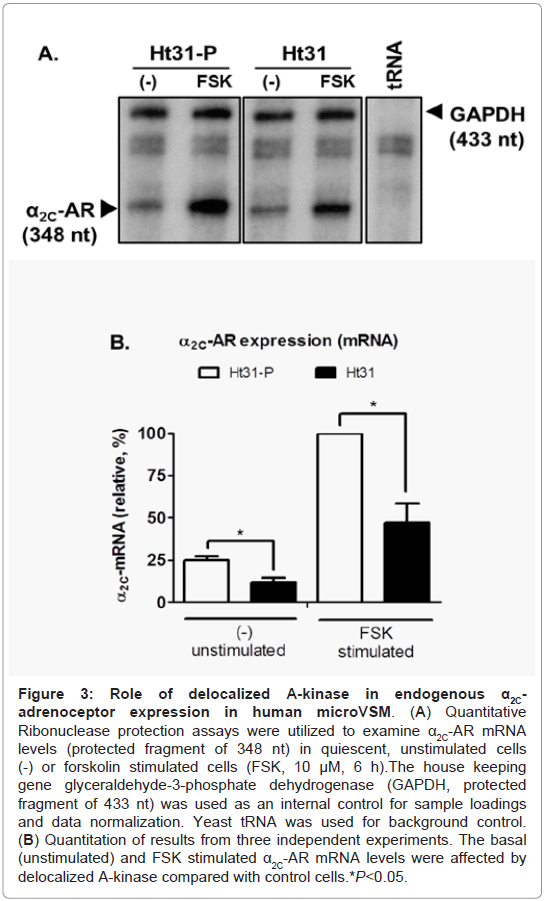
Figure 3: Role of de-localized A-kinase in endogenous α2C-adrenoceptor expression in human microVSM. (A) Quantitative Ribonuclease protection assays were utilized to examine α2C-AR mRNA levels (protected fragment of 348 nt) in quiescent, unstimulated cells (-) or forskolin stimulated cells (FSK, 10 μM, 6 h).The house keeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH, protected ragment of 433 nt) was used as an internal control for sample loadings and data normalization. Yeast tRNA was used for background control. (B) Quantitation of results from three independent experiments. The basal (unstimulated) and FSK stimulated α2C-AR mRNA levels were affected by delocalized A-kinase compared with control cells *P<0.05.
Similarly, imaging studies were performed to assess the effect of Ht31 on α2C-ARs, F-actin, and phospho-Ser2113 filamin-2 in human microVSM stimulated with forskolin (0, 9, and 16 h, Figure 4). In both Ht31-P and Ht31 unstimulated cells (0 h), the α2C-ARs were perinuclear (Figure 4A). However, upon forskolin stimulation (9 h and 16 h) α2C-ARs were on filaments and the cell boundary and colocalized with F-actin (Figure 4B, Figure 4C). Quantitation of α2C-ARs on the cell boundary showed an increase in receptors upon forskolin stimulation in cells transduced with Ht31-P. This increase however, was significantly reduced in cells transduced with Ht31 (Figure 4A, Figure 4B, Figure 4C, and Figure 4D). Quantitation of α2C-ARs in the perinuclear region was also performed. The analysis showed similar reduction of α2C-ARs in cells transduced with Ht31 versus Ht31-P (Figure 4E).Quantitation of changes in F-actin showed a significant increase in Ht31-P transduced cells upon forskolin stimulation at 9 h and 16 h compared with 0 h. However, a significant increase was observed in F-actin at 0 h in cells transduced with Ht31 versus Ht31-P. There was no significant difference at 9 h and 16 h of forskolin stimulation between Ht31 and Ht31-P transduced cells (Figure 4A, Figure 4B, Figure 4C and Figure 4F).
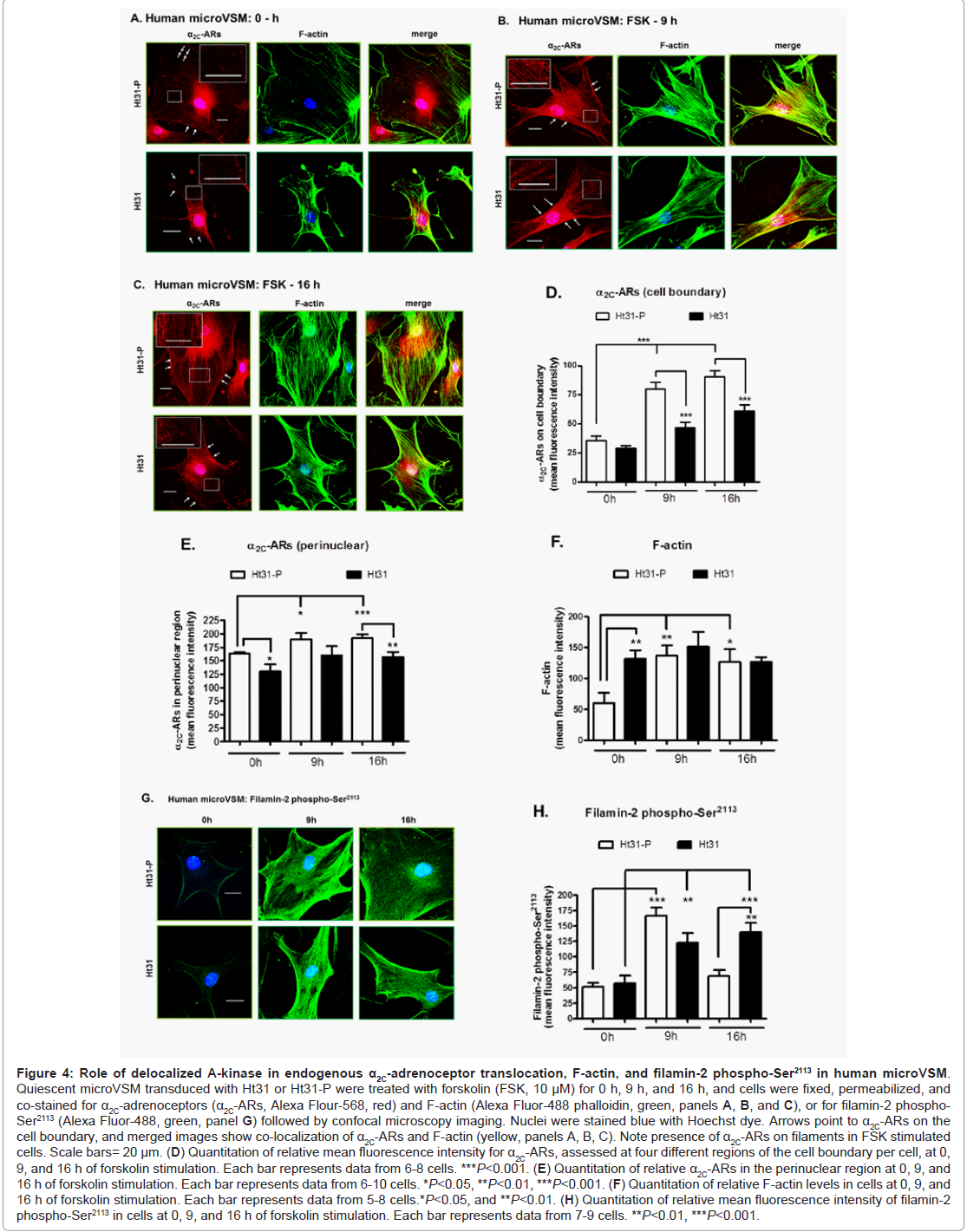
Figure 4: Role of de-localized A-kinase in endogenous α2C-adrenoceptor translocation, F-actin, and filamin-2 phospho-Ser2113 in human microVSM.Quiescent microVSM transduced with Ht31 or Ht31-P were treated with forskolin (FSK, 10 μM) for 0 h, 9 h, and 16 h, and cells were fixed, permeabilized, and co-stained for α2C-adrenoceptors (α2C-ARs, Alexa Flour-568, red) and F-actin (Alexa Fluor-488 phalloidin, green, panels A, B, and C), or for filamin-2 phospho-Ser2113 (Alexa Fluor-488, green, panel G) followed by confocal microscopy imaging. Nuclei were stained blue with Hoechst dye. Arrows point to α2C-ARs on the cell boundary, and merged images show co-localization of α2C-ARs and F-actin (yellow, panels A, B, C). Note presence of α2C-ARs on filaments in FSK stimulated cells. Scale bars= 20 μm. (D) Quantitation of relative mean fluorescence intensity for α2C-ARs, assessed at four different regions of the cell boundary per cell, at 0, 9, and 16 h of forskolin stimulation. Each bar represents data from 6-8 cells. ***P<0.001. (E) Quantitation of relative α2C-ARs in the perinuclear region at 0, 9, and 16 h of forskolin stimulation. Each bar represents data from 6-10 cells. *P<0.05, **P<0.01, ***P<0.001. (F) Quantitation of relative F-actin levels in cells at 0, 9, and 16 h of forskolin stimulation. Each bar represents data from 5-8 cells.*P<0.05, and **P<0.01. (H) Quantitation of relative mean fluorescence intensity of filamin-2 phospho-Ser2113 in cells at 0, 9, and 16 h of forskolin stimulation. Each bar represents data from 7-9 cells. **P<0.01, ***P<0.001.
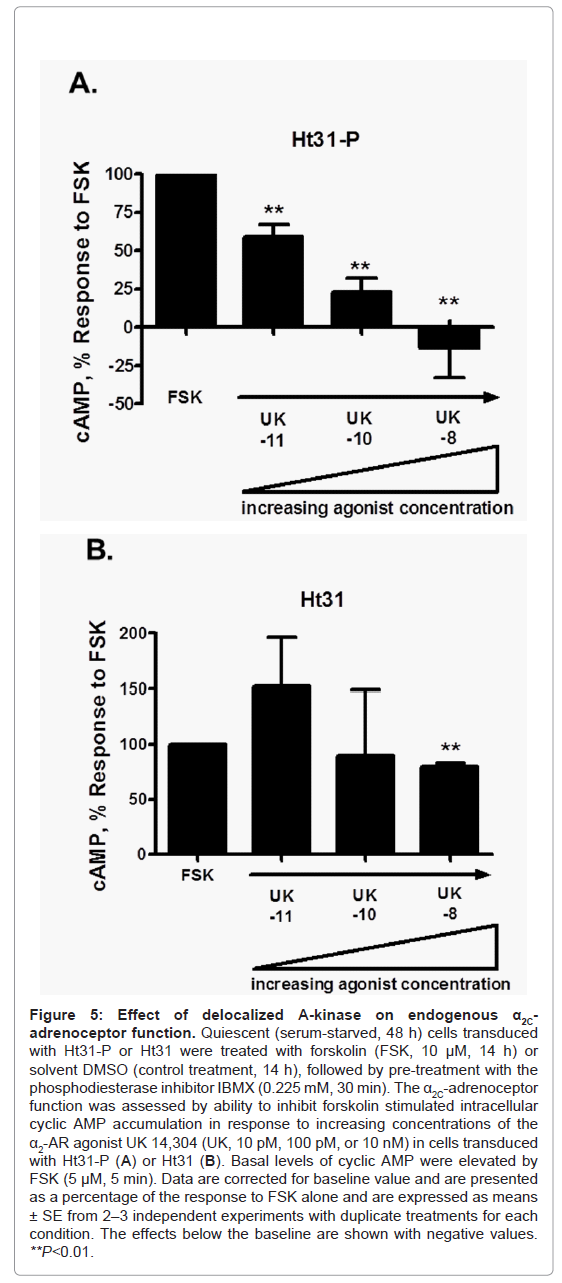
Figure 5: Effect of de-localized A-kinase on endogenous α2C-adrenoceptor function. Quiescent (serum-starved, 48 h) cells transduced with Ht31-P or Ht31 were treated with forskolin (FSK, 10 μM, 14 h) or solvent DMSO (control treatment, 14 h), followed by pre-treatment with the phosphodiesterase inhibitor IBMX (0.225 mM, 30 min). The α2C-adrenoceptor function was assessed by ability to inhibit forskolin stimulated intracellular cyclic AMP accumulation in response to increasing concentrations of the α2-AR agonist UK 14,304 (UK, 10 pM, 100 pM, or 10 nM) in cells transduced with Ht31-P (A) or Ht31 (B). Basal levels of cyclic AMP were elevated by FSK (5 μM, 5 min). Data are corrected for baseline value and are presented as a percentage of the response to FSK alone and are expressed as means ± SE from 2–3 independent experiments with duplicate treatments for each condition. The effects below the baseline are shown with negative values. **P<0.01.
Discussion
Cyclic AMP has a vital role in blood vessel physiology, particularly intracellular signaling through the classic A-kinase, and the more recently identified, Epac-Rap1 pathways. It can lead to dilation of large blood vessels and constriction of small blood vessels, including arterioles. Cyclic AMP can therefore lead to modulation of blood vessel diameter and consequently blood flow to tissues and organs. Identification of the molecular targets of cAMP is therefore critical for a better understanding of the physiological mechanisms involved.In this study we examined and identified mechanism of cAMP signaling in human microVSM derived from cutaneous arterioles, and provide evidence of a physiological role of compartmentalized cAMP-A-kinase signaling using the G protein coupled α2C-ARs as an example. Indeed, these microVSM have served as a unique model in previous studies to elucidate mechanisms of expression and trafficking of endogenous α2C-ARs [3-6,10,30]. Cyclic AMP has a unique role in these microVSM, leading to increased expression and cell surface trafficking of functional α2C-ARs that mediate vasoconstriction [3,5]. The cAMP-Epac-Rap1A pathway, and not the A-kinase pathway, mediated these effects. The results from the present study show that delocalization of A-kinase has an inhibitory effect on α2C-AR expression. The α2C-AR specific antibody used in the studies recognizes the ER-Golgi processed, mature form of the receptor, shown in earlier studies as the main form involved in receptor translocation [8-11]. The studies therefore suggest that receptor post-translational modifications were not affected, and that delocalized A-kinase targets nuclear signaling, inhibiting α2C-ARs at the transcriptional level. Less receptor is produced and therefore, less receptor is available for translocation. Indeed, previous studies in human microVSM using transient transfections showed that delocalized A-kinase catalytic (C) subunit has the potential to inhibit α2C-AR transcription. The specificity of A-kinase signaling is by-passed by expression of the C subunit in transient transfections, leading to transcriptional inhibition of α2C-AR-promoter driven luciferase reporter [4]. These results suggested that A-kinase has the potential to attenuate α2C-AR expression in microVSM. However, post-transcriptional or combination of transcriptional and post transcriptional mechanisms may be involved and cannot be excluded from the present studies utilizing Ht31 to delocalize endogenous A-kinase.
Since surface receptors determine the sensitivity of response to agonist, these results suggest that delocalized A-kinase may affect strength of receptor biological response, reducing α2C-AR mediated physiological response of arteriole constriction. Indeed, this model is supported by functional studies in microVSM that showed attenuated receptor biological responses with Ht31 compared with Ht31-P. A limitation of the study is that it did not test A-kinase inhibitors to reverse the effect on α2C-ARs. However, despite this limitation, these studies suggest that compartmentalized cAMP signaling elicits a selective, and protective, cellular response to maintain microVSM function, which may have relevance to arteriole (or resistance vessel) physiological function.
A possible mechanism mediating this effect may involve phosphorylation of A-kinase substrates linked to α2C-AR regulation; for example, A-kinase may modulate Rap1A and RhoA biological activity. In non-VSM A-kinase phosphorylation at Ser180 can potentially alter biological function of Rap1A by impairing ability to associate with effector proteins [31-35], whereas A-kinase can inhibit RhoA activity by phosphorylation at Ser188, affecting actin stressfiber formation and focal adhesion complexes [36-39]. A-kinase can therefore negatively regulate key molecules that are essential for α2C-AR expression, cytoskeletal organization, and receptor translocation, reducing α2C-ARs, F-actin, and phospho-Ser2113 filamin-2. Although reduced α2C-ARs were noted, the studies however did not show reduced phospho-Ser2113 filamin-2, necessary for receptor translocation. Increased phospho-Ser2113 filamin-2 was noted in 9 h and 16 h stimulated cells transduced with Ht31.
Altered distribution or activity of A-kinase can play a role in disease processes including breast cancer cell invasiveness (suppression of Rho activity by A-kinase) [40], heart failure (hyperphosphorylation of calcium release channel by A-kinase, or reduced AKAP-A-kinase interactions) [23,25], and vascular intimal lesions [41]. The biological consequence of disruption of AKAP-A-kinase interactions in microVSM is not well understood. The model system identified in these studies can be used in future studies to examine the consequence of A-kinase disruption of normal homeostatic mechanisms, providing a paradigm for aberrant regulation, potentially in disease states; for example, examining alterations in gene expression profile. Further, over-activity of α2C-ARs has been implicated in peripheral vascular disorder, including primary and secondary Raynaud′s phenomenon, and peripheral vascular condition of cold feet and cold hands [42,43]. Identification of the exact mechanism of A-kinase inhibition of α2C- ARs in human microVSM in this model system may provide novel insights and target(s) to selectively inhibit α2C-ARs, with potential clinical relevance.
Acknowledgements
This study was supported by the Ohio State University Davis Heart & Lung Research Institute and The Research Institute at Nationwide Children’s Hospital (Institutional support to MAC), a graduate training Joint Supervision Program supported by The Egyptian Cultural and Educational Bureau, Washington DC, and Egyptian Cultural and Educational Sector, Cairo, Egypt (awarded to HKBM), and by a National Institutes of Health (NIH) grant HL085487 (BKM).References
- Jeyaraj SC, Unger NT, Chotani MA (2011) Rap1 GTPases: an emerging role in the cardiovasculature. Life Sci 88: 645-652.
- Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, et al. (2011) The cAMPresponsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem 286: 16681-16692.
- Jeyaraj SC, Unger NT, Eid AH, Mitra S, Paul El-Dahdah N, et al. (2012) Cyclic AMP-Rap1A signaling activates RhoA to induce α2C-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells. Am J Physiol Cell Physiol 303: C499-511.
- Chotani MA, Mitra S, Eid AH, Han SA, Flavahan NA (2005) Distinct cAMP signaling pathways differentially regulate α2C-adrenoceptor expression: role in serum induction in human arteriolar smooth muscle cells. Am J Physiol Heart Circ Physiol 288: H69-76.
- Motawea HK, Jeyaraj SC, Eid AH, Mitra S, Unger NT, et al. (2013) Cyclic AMP-Rap1A signaling mediates cell surface translocation of microvascular smooth muscle α2C-adrenoceptors through the actin-binding protein filamin-2. Am J Physiol Cell Physiol 305: C829-C845.
- Eid AH, Chotani MA, Mitra S, Miller TJ, Flavahan NA (2008) Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle α2C-adrenoceptors. Am J Physiol Heart Circ Physiol 295: H266-272.
- Shepard JT, Vanhoutte PM (1979) The human cardiovascular system: facts and concepts: Raven Press, New York.
- Bailey SR, Eid AH, Mitra S, Flavahan S, Flavahan NA (2004) Rho kinase mediates cold-induced constriction of cutaneous arteries: role of α2C-adrenoceptor translocation. Circ Res 94: 1367-1374.
- Chotani MA, Flavahan NA (2011) Intracellular α2C-adrenoceptors: storage depot, stunted development or signaling domain? Biochim Biophys Acta 1813: 1495-1503.
- Chotani MA, Mitra S, Su BY, Flavahan S, Eid AH, et al. (2004) Regulation of α2-adrenoceptors in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 286: H59-67.
- Jeyaraj SC, Chotani MA, Mitra S, Gregg HE, Flavahan NA, et al. (2001) Cooling evokes redistribution of α2C-adrenoceptors from Golgi to plasma membrane in transfected human embryonic kidney 293 cells. Mol Pharmacol 60: 1195-1200.
- Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, et al. (2001) AKAP-mediated targeting of protein kinase a regulates contractility in cardiac myocytes. Circ Res 88: 291-297.
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, et al. (1994) Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature 368: 853-856.
- Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD (1992) Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem 267: 13376-13382.
- Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, et al. (2001) A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J 20: 1651-1662.
- McConnell BK, Popovic Z, Mal N, Lee K, Bautista J, et al. (2009) Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J Biol Chem 284: 1583-1592.
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, et al. (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185-196.
- Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K (2000) Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol 298: 329-339.
- Hayabuchi Y, Dart C, Standen NB (2001) Evidence for involvement of A-kinase anchoring protein in activation of rat arterial KATP channels by protein kinase A. J Physiol 536: 421-427.
- Yang S, Fletcher WH, Johnson DA (1995) Regulation of cAMP-dependent protein kinase: enzyme activation without dissociation. Biochemistry 34: 6267-6271.
- Chotani MA, Chiu IM (1997) Differential regulation of human fibroblast growth factor 1 transcripts provides a distinct mechanism of cell-specific growth factor expression. Cell Growth Differ 8: 999-1013.
- Michel JJ, Scott JD (2002) AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol 42: 235-257.
- Zakhary DR, Moravec CS, Bond M (2000) Regulation of PKA binding to AKAPs in the heart: alterations in human heart failure. Circulation 101: 1459-1464.
- Kapiloff MS, Schillace RV, Westphal AM, Scott JD (1999) mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci 112 ( Pt 16): 2725-2736.
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, et al. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365-376.
- Ruehr ML, Russell MA, Ferguson DG, Bhat M, Ma J, et al. (2003) Targeting of protein kinase A by muscle A kinase-anchoring protein (mAKAP) regulates phosphorylation and function of the skeletal muscle ryanodine receptor. J Biol Chem 278: 24831-24836.
- Feliciello A, Gallo A, Mele E, Porcellini A, Troncone G, et al. (2000) The localization and activity of cAMP-dependent protein kinase affect cell cycle progression in thyroid cells. J Biol Chem 275: 303-311.
- Rubin CS (1994) A kinase anchor proteins and the intracellular targeting of signals carried by cyclic AMP. Biochim Biophys Acta 1224: 467-479.
- Guimaraes S, Moura D (2001) Vascular adrenoceptors: an update. Pharmacol Rev 53: 319-356.
- Eid AH, Maiti K, Mitra S, Chotani MA, Flavahan S, et al. (2007) Estrogen increases smooth muscle expression of α2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol 293: H1955-1961.
- Quilliam LA, Mueller H, Bohl BP, Prossnitz V, Sklar LA, et al. (1991) Rap1A is a substrate for cyclic AMP-dependent protein kinase in human neutrophils. J Immunol 147: 1628-1635.
- Bokoch GM, Quilliam LA, Bohl BP, Jesaitis AJ, Quinn MT (1991) Inhibition of Rap1A binding to cytochrome b558 of NADPH oxidase by phosphorylation of Rap1A. Science 254: 1794-1796.
- Tsygankova OM, Saavedra A, Rebhun JF, Quilliam LA, Meinkoth JL (2001) Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol Cell Biol 21: 1921-1929.
- Hu CD, Kariya K, Okada T, Qi X, Song C, et al. (1999) Effect of phosphorylation on activities of Rap1A to interact with Raf-1 and to suppress Ras-dependent Raf-1 activation. J Biol Chem 274: 48-51.
- Lerosey I, Pizon V, Tavitian A, de Gunzburg J (1991) The cAMP-dependent protein kinase phosphorylates the rap1 protein in vitro as well as in intact fibroblasts, but not the closely related rap2 protein. Biochemi Biophys Res Commun 175: 430-436.
- Ellerbroek SM, Wennerberg K, Burridge K (2003) Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 278: 19023-19031.
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, et al. (1996) Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 15: 510-519.
- Dong JM, Leung T, Manser E, Lim L (1998) cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKα. J Biol Chem 273: 22554-22562.
- Forget MA, Desrosiers RR, Gingras D, Beliveau R (2002) Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J 361: 243-254.
- Cardone RA, Bagorda A, Bellizzi A, Busco G, Guerra L, et al. (2005) Protein kinase A gating of a pseudopodial-located RhoA/ROCK/p38/NHE1 signal module regulates invasion in breast cancer cell lines. Mol Biol Cell 16: 3117-3127.
- Indolfi C, Stabile E, Coppola C, Gallo A, Perrino C, et al. (2001) Membrane-bound protein kinase A inhibits smooth muscle cell proliferation in vitro and in vivo by amplifying cAMP-protein kinase A signals. Circ Res 88: 319-324.
- Michel MC, Insel PA (2012) Can you blame cold feet on Epac (and Rap1A)? Focus on "Cyclic AMP-Rap1A signaling activates RhoA to induce α2C-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells". Am J Physiol Cell Physiol 303: C488-489.
- Flavahan NA, Flavahan S, Mitra S, Chotani MA (2003) The vasculopathy of Raynaud's phenomenon and scleroderma. Rheum Dis Clin North Am 29: 275-291.