
Journal of Clinical and Investigative Dermatology
Download PDF
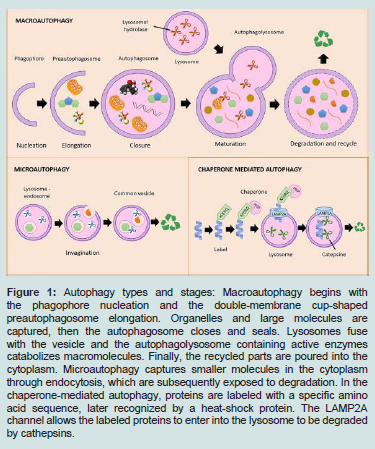 figure 1: Autophagy types and stages: Macroautophagy begins with the phagophore nucleation and the double-membrane cup-shaped preautophagosome elongation. Organelles and large molecules are captured, then the autophagosome closes and seals. Lysosomes fuse with the vesicle and the autophagolysosome containing active enzymes catabolizes macromolecules. Finally, the recycled parts are poured into the cytoplasm. Microautophagy captures smaller molecules in the cytoplasm through endocytosis, which are subsequently exposed to degradation. In the chaperone-mediated autophagy, proteins are labeled with a specific amino acid sequence, later recognized by a heat-shock protein. The LAMP2A channel allows the labeled proteins to enter into the lysosome to be degraded by cathepsins.
figure 1: Autophagy types and stages: Macroautophagy begins with the phagophore nucleation and the double-membrane cup-shaped preautophagosome elongation. Organelles and large molecules are captured, then the autophagosome closes and seals. Lysosomes fuse with the vesicle and the autophagolysosome containing active enzymes catabolizes macromolecules. Finally, the recycled parts are poured into the cytoplasm. Microautophagy captures smaller molecules in the cytoplasm through endocytosis, which are subsequently exposed to degradation. In the chaperone-mediated autophagy, proteins are labeled with a specific amino acid sequence, later recognized by a heat-shock protein. The LAMP2A channel allows the labeled proteins to enter into the lysosome to be degraded by cathepsins.
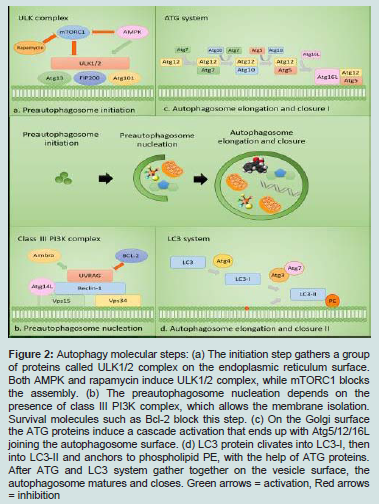 figure 2: Autophagy molecular steps: (a) The initiation step gathers a group of proteins called ULK1/2 complex on the endoplasmic reticulum surface. Both AMPK and rapamycin induce ULK1/2 complex, while mTORC1 blocks the assembly. (b) The preautophagosome nucleation depends on the presence of class III PI3K complex, which allows the membrane isolation. Survival molecules such as Bcl-2 block this step. (c) On the Golgi surface the ATG proteins induce a cascade activation that ends up with Atg5/12/16L joining the autophagosome surface. (d) LC3 protein clivates into LC3-I, then into LC3-II and anchors to phospholipid PE, with the help of ATG proteins. After ATG and LC3 system gather together on the vesicle surface, the autophagosome matures and closes. Green arrows = activation, Red arrows = inhibition
figure 2: Autophagy molecular steps: (a) The initiation step gathers a group of proteins called ULK1/2 complex on the endoplasmic reticulum surface. Both AMPK and rapamycin induce ULK1/2 complex, while mTORC1 blocks the assembly. (b) The preautophagosome nucleation depends on the presence of class III PI3K complex, which allows the membrane isolation. Survival molecules such as Bcl-2 block this step. (c) On the Golgi surface the ATG proteins induce a cascade activation that ends up with Atg5/12/16L joining the autophagosome surface. (d) LC3 protein clivates into LC3-I, then into LC3-II and anchors to phospholipid PE, with the help of ATG proteins. After ATG and LC3 system gather together on the vesicle surface, the autophagosome matures and closes. Green arrows = activation, Red arrows = inhibition
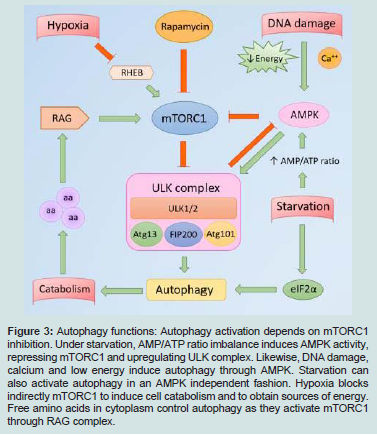 figure 3: Autophagy functions: Autophagy activation depends on mTORC1 inhibition. Under starvation, AMP/ATP ratio imbalance induces AMPK activity, repressing mTORC1 and upregulating ULK complex. Likewise, DNA damage, calcium and low energy induce autophagy through AMPK. Starvation can also activate autophagy in an AMPK independent fashion. Hypoxia blocks indirectly mTORC1 to induce cell catabolism and to obtain sources of energy. Free amino acids in cytoplasm control autophagy as they activate mTORC1 through RAG complex.
figure 3: Autophagy functions: Autophagy activation depends on mTORC1 inhibition. Under starvation, AMP/ATP ratio imbalance induces AMPK activity, repressing mTORC1 and upregulating ULK complex. Likewise, DNA damage, calcium and low energy induce autophagy through AMPK. Starvation can also activate autophagy in an AMPK independent fashion. Hypoxia blocks indirectly mTORC1 to induce cell catabolism and to obtain sources of energy. Free amino acids in cytoplasm control autophagy as they activate mTORC1 through RAG complex.
Review Article
Autophagy: Eating Myself as a Requirement for the Skin Health
Adriana Raquel Rinflerch1*, David Aldo De Luca2, Marcos Mateo Miretti3 and Ricardo Luis Galimberti4
- 1Department of Dermatology, Coordinator of Experimental Dermatology,Italian Hospital of Buenos Aires, Argentina
- 2Department of Dermatology, Italian Hospital of Buenos Aires, Argentina
- 3Graduate in Genetics, Immunogenetics, National University of Misiones;Research Scientist at CONICET, GIGA, IBS, UNaM – CONICET, VacationSpecials, Argentina
- 4Department of Dermatology, UBA Argentina, Italian Hospital of BuenosAires, Argentina
*Address for Correspondence: Adriana Rinflerch, Department of Dermatology, Coordinator of ExperimentalDermatology, Italian Hospital of Buenos Aires, Argentina, Tel: +54 11 49590200; Ext: 5355; E-mail:adriana.rinflerch@hospitalitaliano.org.ar
Citation: Rinflerch AR, De Luca DA, Miretti MM, Galimberti RL. Autophagy: Eating Myself as a Requirement for the Skin Health. J Clin Investigat Dermatol.2017;5(2): 1
Copyright: © 2017 Rinflerch AR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & InvestigativeDermatology| ISSN: 2373-1044 | Volume: 5, Issue: 2
Submission:14 June, 2017| Accepted: 15 July, 2017 | Published: 02 August, 2017
Submission:14 June, 2017| Accepted: 15 July, 2017 | Published: 02 August, 2017
Abstract
Cells recycle their components during starvation through aprocess called autophagy. It is a normal cellular mechanism related tophysiological actions such as nutrient and energy metabolism.
In the skin as in other organs, one normal function of autophagyis related to the immune system as the control of mycotic, bacterialand viral infections. Genetic alterations cause amino acid substitutionsin proteins, when it’s involved to autophagy compounds, results inalterations of the defense mechanisms of the host. They have beenrecently involved in the pathogenesis of immunological disorderssuch as systemic lupus erythematosus, vitiligo and oral lichen planus.Furthermore, alterations in autophagy process induce a moreaggressive behavior and reduced life expectancy in squamouscell carcinoma and malignant melanoma. The aim of this review isdescribe the molecular process of autophagy and how alterations ofcomponents of this process are linked to the development of diversesskin pathologies.
The understanding of molecular mechanisms involved inautophagy promises new diagnostic and treatment perspectives,mainly in autoimmune diseases and skin cancer.
Keywords
Autophagy; Skin; Lupus; Vitiligo; Oral lichen planus; Autoimmune
Abbreviations
HSC70: Heat Shock Chaperone 70 kDa; LAMP2A: Lysosome- Associated Membrane Protein 2; ATG: Autophagy-Related Protein Gene; UNC-51: Uncoordinated Serine/Threonine-Protein Kinase; PI3K III: Phosphoinositol 3 Kinase Class III; LC3: Microtubule- Associated Protein 1 Light Chain 3; ULK1: Unc-51 Like Autophagy Activating Kinase 1; FIP200: FAK Family Kinase-Interacting Protein of 200 kDa; VPS34: Vacuolar Protein Sorting 34; PE: Phosphatidylethanolamine; mTORC1: Mechanistic Target of Rapamycin Complex 1; AMP: Adenosine Monophosphate; AMPK: AMP-Activated Protein Kinase; UVRAG: UV Radiation Resistance- Associated Gene Protein; AMBRA1: Autophagy and Beclin 1 Regulator 1; Bcl-2: B cell lymphoma 2; TYR: Tyrosinase; MITF: Microphthalmia-Associated Transcription Factor; EIF2α: Eukaryotic Translation Initiation Factor 2 Subunit α; RHEB: Ras Homolog Enriched In Brain; HSV: Herpes Simplex Virus; SLE: Systemic Erythematous Lupus; NSG: Non Segmental Vitiligo; VS: Segmental Vitiligo; OLP: Oral Lichen Planus; HGS: Hepatocyte Growth Factor- Regulated Tyrosine Kinase Substrate; ESR1: Estrogen Receptor 1; SNCA: Synuclein Alpha; SCC: Squamous Cell Carcinoma; 5-FU: 5-Fluorouracil; MM: Malignant Melanoma
Introduction
Autophagy is a catabolic reaction carried out by lysosomal enzymes. Under starvation conditions, the cell recycles and consumes itself, through a process that is equivalent in plant, animals and fungi cells [1-3]. This cannibalization plays a fundamental role in the use of nutrients, oxygen and energy, along with the replacement of damaged or aged organelles [4-5].
Under homeostatic environment, autophagy functions as an internal quality control and as cell defense by degrading dysfunctional proteins, organelles, as well as bacterial or viral components. In addition, autophagy is involved in immune response as it processes not only the self-proteins as self-antigens, but also the foreign ones to eliminate the trigger antiviral immune response. Under cell differentiation, autophagy induces specific gene expression: In basal state, autophagy blocks apoptosis while on the other hand itaccelerates carcinogenesis [6-8].
Investigations to understand the mechanism of autophagy were carried out in yeast, and their human counterparts were then discovered. The preservation of the functionality of these proteins indicates that this process is elementary and was acquired very early in the evolution of living beings [2,3].
There are two recycling mechanisms of cell components. One of them addresses the short half-life proteins degradation, mediated by the ubiquitin system. The second one called autophagy, removes long half-life proteins and also organelles [9,10].
This review aims to introduce autophagy as a physiological mechanism that recycles organelles, cleans the cytoplasm and activates immune response. Furthermore, we overview the effects of mutation in autophagy pathways in skin disorders.
Types of autophagy
When a peptide or organelle is destined to be degraded, the cell can use different alternatives. One of them is known as chaperonemediated autophagy, where a group of amino acids is recognized at the amino terminal end of the protein to be degraded and transported to the lysosome. Alternatively, there is the microautophagy in whichendosomes participate, and macroautophagy, a process in which a double membrane system is triggered. Autophagy types are illustrated in Figure 1 [5,9,11].
Chaperone-mediated: Polypeptides to be split are labeled by five amino acids (KFERQ or Lys-Phe-Glu-Arg-Gln) at the amino end. The KFERQ tag is recognized by chaperone proteins called Heat shock chaperone 70 kDa (HSC70) and transferred to the lysosome by Lysosome-associated membrane protein 2 (LAMP2A) [7,10].
Microautophagy: The material to be degraded locates within endosome(s). Both lysosome and endosome membranes fuse and the common vesicle contains active enzyme such as cathepsins to achieve proteolysis [7].
Macroautophagy: It consists of a series of dynamic membrane events in the cytoplasm. Macroautophagy begins with the sac nucleation, a double membrane cup-shaped structure called preautophagosome that surrounds from proteins to entire organelles. Once the material is wrapped, the double membrane closes and seals and becomes an autophagosome. Subsequently it fuses with the lysosome, which stores lipases and proteases in low pH and forms the phagolysosome. The enzymes dissociate intermolecular junctions, solubilizes their components, return them to the cytoplasm to be recycled [5,7,12].
Molecular processes
The autophagosome has a half-life of approximately 10 minutes before it merges with the lysosome and loses its identity. The phagosome lipids come mainly from the Endoplasmatic Reticulum (ER) and the Golgi complex, and their inner membrane containspoorly or no proteins, while the outer membrane includes more than 35 proteins revealing a highly specialized structure [3].
Genes related to the autophagy process are called Autophagy- Related Protein (ATG). Some have exclusive functions, like the ATG12 conjugation system discovered by Dr. Mizushima, who was the first to identify ubiquitin-like-system proteins, while others are common to other cellular processes [3,6,11,13].
The molecular machinery involved in autophagy consists of four functional groups: The kinase complex Uncoordinated Serine/ Threonine-Protein Kinase (UNC-51), class III PI3K complex and two ubiquitin-like complexes: ATG5-ATG12 and Microtubule-associated protein 1 Light Chain 3 (LC3) - Phosphatidylethanolamine (PE) system also called LC3-II [2,7].
The first stages of the autophagy are the preautophagosome nucleation and elongation stages, followed by its fusion with the lysosome and the genesis of the autophagolysosome. The autophagy steps are described below and shown on Figure 2.
Preautophagosome initiation
Preautophagosome starts to shape when groups of proteins kinase-like UNC-51, including ULK1 or ULK2, ATG13, FAK family kinase-interacting protein of 200 kDa (FIP200) and ATG101 gather together and form a protein complex [2,7].
Nucleation
In this step, a group of proteins and phospholipids are recruitedto form the Preautophagosome. Nucleation requires PI3K class III complex, composed of Vacuolar protein sorting 34 (VPS34), PI3K and the regulatory subunit ATG14L, VPS15 and Beclin 1 also known as ATG6 [2,3,7].
Elongation
The expansion and closure of the preautophagosome membrane require two ubiquitin-like pathways conjugation. The first attaches ATG5-ATG12 with ATG16L, while the second results in the conjugation of PE to LC3 also known as ATG8. After recognizing the material to degrade, the autophagosome membrane binds the lysosome, due to the conjugation of PE and LC3. Once the autophagosome fuses with the endocytic and lysosomal components, it becomes an autolysosome [2,3,7].
Regulation
Every autophagy step is under a fine control. As an example, both mTORC1 and rapamycin are capable of inhibiting the activity of ULK1 and therefore, the initiation of autophagy. Other proteins like AMPK pivots under the influence of AMP levels. AMPK contributes to the inhibition exerted by Mammalian Target of Rapamycin Complex 1 (mTORC1) or it can phosphorylate ULK1 when activation of the autophagy process is required [2,3,7].
In the nucleation stage, there are different regulatory points. Whereas UV Radiation Resistance-Associated Gene Protein (UVRAG) and Autophagy and Beclin 1 Regulator 1 (AMBRA1) participate positively on autophagosome nucleation, B Cell Lymphoma 2 (Bcl-2) and survivin have a negative effect. In the case of AMBRA1, the protein binds Beclin-1 and Bcl-2 to trigger autophagy. The last checkpoint is LC3 cleavage by ATG enzymes ATG7 and ATG3. After LC3 proteolysis, LC3-I binds PE to give rise to LC3-I I [1,14]
Autophagy Functions
Autophagy regulates the energy and protein consumption. Under starvation the Adenosine Monophosphate (AMP)/ Adenosine Triphosphate (ATP) cell ratio increases, AMP-Activated Protein Kinase (AMPK) triggers autophagy through the ULK complex activation. At the same time, starvation induces EIF2α (eukaryotic translation initiation factor 2 subunit α) expression and therefore, autophagy and protein catabolism. The availability of amino acids in the cytoplasm stimulates RAG, which leads to mTORC1 activation, one of the central autophagy gatekeepers. Other stimuli like DNA damage, low energy and calcium enhance AMPK activity, which blocks mTORC1. RHEB is a GTPase that binds and activates mTORC1 to induce protein synthesis. Low oxygen tension inhibits RHEB, therefore, inactivates mTORC1 and protein anabolism. Indirectly, low oxygen enhances eIF2α phosphorylation and releases autophagy from inhibition (Figure 3) [2,3,8].
In the skin, as well as in other organs, autophagy activates the immune system to control infections. However, it also contributes to autoimmune diseases, photodamage and cell senescence under certain conditions as detailed below.
In the skin, as well as in other organs, autophagy activates the immune system to control infections. However, it also contributes to autoimmune diseases, photodamage and cell senescence under certain conditions as detailed below.
Autophagy in dermatology
Autophagy enhances bacterial and yeast immune responsein skin diseases, although it may control viral infections or permit virus replication based on the level of expression of autophagy. The UV light inactivates autophagy and leads to cellular senescence. In certain autoimmune diseases, the effect of autophagy is ambiguous. In systemic lupus erythematosus, enhanced autophagy correlates with T and B cell activity, therefore, increased disease activity while on the other hand, and in vitiligo expresses melanogenesis and pigmentation. Oral lichen planus develops an autophagy blockade that induces epithelial apoptosis. In skin cancer such as melanoma and squamous cell carcinoma, they show an active autophagy, which correspond to tumor progression, metastasis and therapy resistance.
Skin infections:Autophagy represents a defensive barrier against pathogens and helps to restore cell homeostasis. During the autophagolysosome fusion, the endosome containing several microorganisms is exposed to lysosomal enzymes. This process leads to pathogens degradation and therefore, autophagy has antimicrobial effects [14].
Autophagy plays a major defensive role against intracellular bacteria. In skin infections such as impetigo and erysipelas, group A Streptococcus evades endocytosis and autophagy by chaperones, but it is vulnerable to macrophagy [15].
Staphylococcus aureus is also susceptible to autophagosome, butcertain bacterial strains carry toxin genes, which provide resistance toautophagy, avoiding lysosome fusion with autophagosome. S. aureususes the endosome as a niche to replicate, and then the infectionspreads from the skin into the blood vessels, causing metastaticdisease [16].
Candida albicans is a yeast capable of producing superficial skin infections such as candidal intertrigo or invasive forms withhematogenous dissemination. Macrophages are in charge of controlling Candida infection through autophagy. Nicola, et al. proved that blocking experimentally ATG5 protein reduced the fungistatic activity of macrophages [17].
The role of autophagy in viral diseases could be either permissive or may control the infection. For example, autophagy has an antiviral effect on Herpes Simplex Virus-1 (HSV-1) as induces virus uptake, degradation and antigen presentation through class I histocompatibility molecules [18]. On the other hand, basal levelof autophagy allows a Herpes Simplex Virus-2 (HSV-2) persistent infection, whereas an active autophagy achieves to control HSV-2 infection. During an active infection, HSV-2 blocks the autophagyinduction but maintains a basal and permissive level for the virus to spread. Yokoub, et al. demonstrated that during the HSV-2 infection, ATG5 protein plays a permissive role and ATG5 blockade confers resistance to the virus [19]. Finally, both herpes virus developed defense mechanisms against autophagy. The ICP34.5 protein confers neurovirulence to HSV-1 and HSV-2 and blocks Beclin-1, that prevents from the autophagosome development [20].
Photodamage and senescence: Ultraviolet A and B rays have the ability to alter not only the keratinocytes DNA, but also to infiltrate into the dermis and reach the fibroblasts. These cells are photosensitive and both UVA and UVB exposure induce a cellular process called cell aging or senescence. This concept includes specific metabolicchanges that inhibit the cellular division as a consequence of thetelomeres shorten and DNA damage [1,21]. Moreover, Pernodet, etal. described a 77.9% reduction in autophagy in aged normal human skin fibroblast, when they measure LC3B expression by RT-PCR. They also found that there is an autophagy nighttime peak only in young fibroblast and absent in aged ones. The low UVA exposure also reactivated autophagy in aged cells [22].
However, molecular events involved in cell senescence by UV exposure and autophagy in fibroblasts are complex. A group of ribonucleic acid molecules called microRNAs are able to control cell proliferation, differentiation, apoptosis and even autophagy [23]. UV light exposure can also activate some microRNAs transcription, particularly miR-23a, which induces cellular senescence indirectly through AMBRA1. As mentioned above, under normal conditions AMBRA1 stimulates autophagy. MiR-23a suppresses AMBRA1, thereby reducing autophagy levels and triggering events of cellular aging [1].
Autoimmune diseases
Autophagy fulfills important functions in the immune response, such as the recognition and presentation of antigens, in addition to the ontogenesis and regulation of the immune system. Autophagy malfunctions are involved in autoimmune disease development. The nature of elicited adaptive immune response is primarily conditioned by the presentation of cytosolic and exogenous phagocytosed protein degraded antigens loaded onto Major Histocompatibility Complex (MHC) class I and class II surface molecules, respectively. Peptide antigens derived from endogenous proteins (cytosolic, nuclear, cytoskeletal and viral protein) have nevertheless been found in MHC class II molecules from B cells [24].
CD4+T cells activation by cytosolic antigens from autophagosomes provides an additional mechanism to trigger an immune response against bacteria and virulence factors escaping into the cytosol. Therefore, macroautophagy ensures the presentation of self and internalized proteins residing in the cytosol of Antigen Presenting Cells (APC), possibly by fusing autophagosomes to MIICs [25]. Self-proteins redirected to the class II antigen processing pathway are essential for CD4+T cell selection in the thymus, assuring a self tolerant T cell repertoire [26]. Failure in B and T cell selection in primary organs, imbalance in central / peripheral tolerance and in the homeostasis of the immune response are major causes of autoimmune disorders [27,28].
Systemic Lupus Erythematosus (SLE): SLE is a chronic autoimmune disease with multisystemic involvement, due to autoreactive lymphocytes. There is also evidence that ATG nucleotide polymorphisms predispose toward triggering lupus and they could induce different severity of SLE [14,29,30].
Regarding CD4+T cells, patients with SLE have constitutively higher levels of autophagy compared to healthy controls. The lymphocyte ontogeny analysis revealed that thymic cells, responsible for immunological tolerance, present a normal level of autophagy. For this reason, T cells homeostasis imbalance appears to be peripheral or extrathymic. In addition, hydroxychloroquine is a medication used for the treatment of SLE and it was observed that by itself, the drug inhibits the autophagy flow. However, the autophagosomes accumulation in CD4+T cells cannot only be explained by the use of hydroxychloroquine as patients treated with other drugs showed the same behavior [31].
Concerning autoantibodies synthesis in SLE, B cells play a fundamental role. In SLE patients, autophagy shows activity levels above normal range in immature B cells, which prevents autoreactive lymphocytes from negative selection and a subsequent apoptosis. On the other hand, antibody synthesis induces a metabolic stress, along with mitochondrial substitution, processes driven by ATG5 and ATG7 gene products, capable of blocking apoptosis. While overexpression of Bcl-2 blocks apoptosis, Bcl-2 suppression in SLE induces autophagy. These molecular events create an autoreactive B cell death resistance environment with a prolonged survival and a sustained production of autoantibodies [32].
Vitiligo: Vitiligo is an autoimmune disease characterized by the absence of mucocutaneous pigment with two major clinical subtypes:Segmental (SV) and Non-Segmental Vitiligo (NSV). In the latter one, achromic macules are classified as generalized, acrofacial or universal vitiligo. Although the differential pathophysiology between the two clinical forms is not fully elucidated, there is a differential expression of certain genes in white blood cells. Wang, et al. confirmed variations in innate immunity, apoptosis and autophagy gene expression. In melanocytes, autophagy acts as a defense mechanism against UVexposure and oxidative stress [33-35].
Nie, et al. analyzed autophagy activity and LC3-PE expression in melanocytes found in vitiligo margins in SV, NSV (generalized variant) patients and healthy skin. Both new autophagosomes and LC3-PE expression were detected without any stimuli in SV and NSV, but not in healthy skin. With the addition of rapamycin, a drug that inhibits mTORC1 and promotes autophagy, autophagosome expression was upregulated in both forms of vitiligo and in healthy skin. Simultaneously, Microphthalmia-Associated TranscriptionFactor (MITF) and Tyrosinase Gene (TYR) melanin-related gene expression were overexpressed during rapamycin autophagy induction. Therefore, autophagy could function as a defense mechanism against vitiligo, as it induces melanogenesis on the lesion margins [36].
Oral Lichen Planus (OLP): Lichen planus is a rare autoimmune process that affects skin, mucosa and skin adnexa. The oral presentation consists of white papules grouped in a linear or reticularmanner and atrophic or erosive forms are also described. T cells arethe major actors that trigger keratinocytes apoptosis [37].
The ATG9 gene encodes membrane proteins linked to autophagy regulation and innate immune inhibition. Tan, et al. found a lower ATG9B expression in OLP compared to healthy controls. ATG9B acts also as an oxygen sensor and prevents from apoptosis. The lower ATG9B expression leads to apoptosis of the oral epithelium, a typical event of lichen planus [38].
Many other genes such as Hepatocyte Growth Factor-Regulated Tyrosine Kinase Substrate (HGS), Estrogen Receptor 1 (ESR1) and Synuclein Alpha (SNCA) were found downregulated in OLP T cells. The proteins encoded are responsible for lysosomal transport, vesicle recycling and autophagosomes metabolism. In addition, autophagy inhibition releases SNCA protein into the extracellular space, with an inflammatory exacerbation and cell damage, events related to OLP [38].
Skin cancer
Autophagy has a pivotal role in skin oncogenesis. On the one hand, it acts as a tumor suppressor in normal cells, prevents from genetic damage accumulation and inhibits mitosis in adverse genomic conditions. Autophagy also induces cell survival in established neoplasms as an adaptive mechanism to overcome hypoxia and cell starvation. Beclin-1 allele deletion was detected in certain cancers (breast, prostate and ovary), which implies the tumor suppressor function loss [14,39-41].
In melanoma and Squamous Cell Carcinoma (SCC), autophagy fulfils the metabolic demand and promotes cell growth and division. Higher autophagic activity correlates with tumor aggression and metastasis, while blocking autophagy expression induces apoptosis [42].
Squamous cell carcinoma:Zhang, et al. confirmed that LC3 expression in non-invasive SCC is reduced, whereas the antiapoptotic Bcl-2 and survivin are overexpressed, unlike normal skin. Claerhout, et al. demonstrated that invasive and metastatic SCC showed high expression of LC3 with low Bcl-2 and survivin levels. The degree of LC3 expression is a prognostic factor associated with tumor thickness, TNM stage, tumor subtype and the presence of lymph node metastases[43-45].
The use of chemotherapeutic drugs such as 5-FU, induces autophagy in metastatic SCC, that leads to an ulterior therapy resistance. The enhanced autophagy in advance SCC is reflected by LC3 overexpression. A new autophagy inhibitor 3-methyladenine helps to surpass the drug resistance. Both 3-MA and 5-Fluorouracil(5-FU) inhibit proliferation and tumor invasion and induce apoptosis in SCC cells. Therefore, autophagy appears to be a new therapeutic target for overcoming drug resistance in advanced SCC [43].
Melanoma:Melanoma is a less frequent, highly mitotic and metabolic skin cancer with greater local and distant metastasis rate. Choi, et al. described that Malignant Melanoma (MM) autophagic activity induces cell survival and a higher chance of metastatic invasion [39].
Lazova, et al. found that isoform B of LC3 protein was detected not only in in-situ and invasive types of MM, but also in tumor-associated macrophages. LC3B protein expression enhances under stress conditions in the Golgi complex and induces the autophagosomes synthesis. Active autophagy enables metabolic conditions for tumor invasion [46]. Pawelek, et al. discovered that autophagy in M provides nutrients from the extracellular space, wrapped in autophagosomes. This behavior was observed in metastatic MM rather than in other neoplasms. The positive metabolic energy balance provides for certain vasculature-independence [47]. Furthermore, MM autophagosomes contain β1-6 branched oligosaccharides bound to integrin and growth factor receptors. They enhance cell migration and metastasis, facts that correlate to a low rate survival in patient with advanced melanoma [46].
The role of autophagy in MM is not fully clarified, as many cell survival mechanisms depend on the environment. For instance, miR-638 blocks p53, a protein that checks the DNA condition before mitosis. Blockade of miR-638 enhances autophagy in vitro, leads to cell growth suppression and also induces apoptosis. For those reasons, autophagy activates intricate and opposed mechanisms, depending on the balance between pro and antiapoptotic factors [48].
Perspectives and Clinical Applications
Some time ago, cell and genetic discoveries were only part of basic research and therefore, destined to biology books. Nevertheless, the countless experimental results, along with the help of translational medicine, enhance the molecular knowledge and allow choosing better therapies for chronic diseases such as diabetes, hypertension and cancer.
Precise mutations in autophagy components were recognized in specific diseases. Based on this statement, valuable biomarkers for molecular diagnostic introduce a new concept to the clinical practice: the personalized medicine. The early identification of these mutations along with suitable treatments, would allow preventing a disease, before it manifests. New advanced therapy tools are under development and gene therapy is not a futuristic illusion anymore.
Conclusion
Autophagy is a tightly coordinated and conserved process among different species, which allows metabolism and energy regulation, cell machinery reparation and immunity functions. As it was mentioned above, many autophagy steps could be altered leading to infections, autoimmune diseases and skin cancer. There are still many questions to answer from a physiological and biochemical point of view to better understand skin diseases and to enable possible therapeutic targets in the future.
References
- Zhang JA, Zhou BR, Xu Y, Chen X, Liu J, et al. (2016) MiR-23a-depressed autophagy is a participant in PUVA- and UVB-induced premature senescence. Oncotarget 7: 37420-37435.
- Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24: 9-23.
- Wesselborg S, Stork B (2015) Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell Mol Life Sci 72: 4721-4757.
- Shintani T, Klionsky DJ (2004) Autophagy in health and disease: A double-edged sword. Science 306: 990-995
- Lodish H, Berk A (2016) Vesicular traffic, secretion and endocytosis. In: CTI Reviews, Molecular cell biology, (7thedn). Cram101 Textbook Reviews, USA, pp. 287.
- White KA, Luo L, Thompson TA, Torres S, Hu CA, et al. (2016) Variants in autophagy-related genes and clinical characteristics in melanoma: A population-based study. Cancer Med 5: 3336-3345.
- Li L, Chen X, Gu H (2016) The signaling involved in autophagy machinery in keratinocytes and therapeutic approaches for skin diseases. Oncotarget 7: 50682-50697.
- Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27-42.
- Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461-472.
- Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: Roles in disease and aging. Cell Res 24: 92-104.
- Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: The interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 5: 81-94.
- Akinduro O, Sully K, Patel A, Robinson DJ, Chikh A, et al. (2016) Constitutive autophagy and nucleophagy during epidermal differentiation. J Invest Dermatol 136: 1460-1470.
- Farré JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat Rev Mol Cell Biol 17: 537-552.
- 14.Yu T, Zuber J, Li J (2015) Targeting autophagy in skin diseases. J Mol Med (Berl) 93: 31-38..
- Barnett TC, Liebl D, Seymour LM, Gillen CM, Lim JY, et al. (2013) The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14: 675-682.
- Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, et al. (2007) Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 282: 2695-2706.
- Nicola AM, Albuquerque P, Martinez LR, Dal-Rosso RA, Saylor C, et al. (2012) Macrophage autophagy in immunity to cryptococcus neoformans and candida albicans. Infect Immun 80: 3065-3076.
- English L, Chemali M, Duron J, Rondeau C, Laplante A, et al. (2009) Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol 10: 480-487.
- Yakoub AM, Shukla D (2015) Basal autophagy is required for herpes simplex virus-2 infection. Sci Rep 5: 12985.
- Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, et al. (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1: 23-35.
- Cavinato M, Koziel R, Romani N, Weinmüllner R, Jenewein B, et al. (2017) UVB-induced senescence of human dermal fibroblasts involves impairment of proteasome and enhanced autophagic activity. J Gerontol A Biol Sci Med Sci 72: 632-639.
- Pernodet N, Dong K, Pelle E (2016) Autophagy in human skin fibroblasts: Comparison between young and aged cells and evaluation of its cellular rhythm and response to ultraviolet A radiation. J Cosmet Sci 67: 13-20.
- David DL, Adriana R (2015) Non-coding ribonucleic acids: Stepping into the era of personalized medicine. Dermatol Argent 21: 254-263.
- Costantino CM, Spooner E, Ploegh HL, Hafler DA (2012) Class II MHC self-antigen presentation in human B and T lymphocytes. PLoS One 7: e29805.
- Münz C (2012) Antigen processing for mhc class ii presentation via autophagy. Front Immunol 3: 9.
- Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L (2008) Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455: 396-400.
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, et al. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90-97.
- Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, et al. (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11: 55-62.
- Liu X, Qin H, Xu J (2016) The role of autophagy in the pathogenesis of systemic lupus erythematosus. Int Immunopharmacol 40: 351-361.
- Zhou XJ, Zhang H (2012) Autophagy in immunity: Implications in etiology of autoimmune/autoinflammatory diseases. Autophagy 8: 1286-1299.
- Gros F, Arnold J, Page N, Décossas M, Korganow AS, et al. (2012) Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 8: 1113-1123.
- Clarke AJ, Ellinghaus U, Cortini A, Cortini A, Stranks A, et al. (2015) Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis 74: 912-920.
- Wang P, Li Y, Nie H, Zhang X, Shao Q, et al. (2016) The changes of gene expression profiling between segmental vitiligo, generalized vitiligo and healthy individual. J Dermatol Sci 84: 40-49.
- Ezzedine K, Lim HW, Suzuki T, Katayama I, Hamzavi I, et al. (2012) Revised classification/nomenclature of vitiligo and related issues: the vitiligo global issues consensus conference. Pigment Cell Melanoma Res 25: E1-E13.
- 35.Qiao Z, Wang X, Xiang L, Zhang C (2016) Dysfunction of autophagy: A possible mechanism involved in the pathogenesis of vitiligo by breaking the redox balance of melanocytes. Oxid Med Cell Longev 2016: 3401570.
- Nie HQ, Wang P, Zhang XY, Ding C, Liu J, et al. (2016) Relationship between autophagy of melanocytes in patients with vitiligo and clinical types. Zhonghua Yi Xue Za Zhi 96: 2064-2069.
- Bascones -Ilundain C, González Moles MA, Carrillo de Albornoz A, Bascones-Martínez A (2006) Oral lichen planus (I). Clinical, etiopathogenic and epidemiological aspects. Av. Odontoestomatol 22: 11-19.
- Tan YQ, Zhang J, Du GF, Lu R, Chen GY, et al. (2016) Altered autophagy-associated genes expression in t cells of oral lichen planus correlated with clinical features. Mediators Inflamm 2016: 4867368..
- Choi KS (2012) Autophagy and cancer. Exp Mol Med 44: 109-120.
- Choi SR, Chung BY, Kim SW, Kim CD, Yun WJ, et al. (2014) Activation of autophagic pathways is related to growth inhibition and senescence in cutaneous squamous cell carcinoma. Exp Dermatol 23: 718-724.
- Gozuacik D, Kimchi A (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23: 2891-2906.
- Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, et al. (2011) Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 17: 3478-3489.
- Zhang L, Zhang J, Chen L, Wang J (2015) Autophagy in human skin squamous cell carcinoma: Inhibition by 3-MA enhances the effect of 5-FU-induced chemotherapy sensitivity. Oncol Rep 34: 3147-3155.
- Yoshihara N, Takagi A, Ueno T, Ikeda S (2014) Inverse correlation between microtubule-associated protein 1A/1B-light chain 3 and p62/sequestosome-1 expression in the progression of cutaneous squamous cell carcinoma. J Dermatol 41: 311-315.
- Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, et al. (2010) Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer 127: 2790-2803.
- Lazova R, Klump V, Pawelek J (2010) Autophagy in cutaneous malignant melanoma. J Cutan Pathol 37: 256-268.
- Pawelek JM (2008) Cancer cell fusion with migratory bone marrow derived cells as an explanation for metastasis: New therapeutic paradigms. Future Oncol 4: 449-452.
- Bhattacharya A, Schmitz U, Raatz Y, Schönherr M, Kottek T, et al. (2015) miR-638 promotes melanoma metastasis and protects melanoma cells from apoptosis and autophagy. Oncotarget 6: 2966-2980.