
Review Article
Mechanisms of Stimulant Drug Dependence
Joseph D. Miller1*and Richard E. Wilcox2
- 1Department of Pharmacology, American University of the Caribbean School of Medicine, USA
- 2Division of Pharmacology and Toxicology, College of Pharmacy, University of Texas at Austin, USA
*Address for Correspondence: Joseph D. Miller, Department of Pharmacology, American University of the Caribbean School of Medicine, St. Maarten, Florida, USA, Tel: 721-545-2298; Fax: 512-475-6088; E-mail: jmiller2@aucmed.edu
Citation: Miller JD, Wilcox RE. Mechanisms of Stimulant Drug Dependence. J Addiction Prevention. 2017;5(1): 6.
Copyright: © 2017 Miller JD, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Addiction & Prevention | ISSN: 2330-2178 | Volume: 5, Issue: 1
Submission: 13 May, 2017 | Accepted: 15 June, 2017 | Published: 23 June, 2017
Keywords:
Drug abuse; Drug dependence; Addiction; Substance use disorder; Genetics of drug dependence; Theories of drug dependence; Treatment of drug dependence; Drugs for craving
Abstract
Objectives: Current theories of severe substance use disorder (chemical dependence) have tended to focus on either the initial phases of the process when the individual experiences WANT or on the later phases when the predominant experience is NEED for the drug. The present review integrates these two phases of the dependence process using information from our own formulation in conjunction with other theories plus selected results from the anti-drug craving literature.
Methods: We have recently provided an integrated theory of severe substance use disorder (dependence) that integrates incentive sensitization, receptor down-regulation, psychomotor stimulant, and opponent process theories into a framework. The literature that helped to derive these theories was integrated with recent publications by these and other authors from searches done on PubMed using search criteria as follows: review article, free full text, published since 2000, and drug addiction or drug dependence.
Results: Persistence of craving implies and neurobiological research has confirmed that chemical dependence changes brain chemistry in those regions of the brain that are associated with emotion, motivation and decision-making (the limbic system). The key parts of the limbic system that are most altered by the chemical dependence process are the mesolimbic/mesocortical dopamine (DA) system and the nigrostriatal DA system. The anatomical substrate of chemical dependence includes specific portions of the DA projections to the forebrain. Significantly, all major theories of drug dependency associate DA in these anatomical pathways with the different stages and effects of drug usage and chemical dependence. We discuss in this paper the relationship between dopamine neuroanatomy and neurophysiology and the well-known drug dependency continuum stretching from LIKE to WANT to NEED.
Conclusions: We suggest that there is a continuum of drug abuse extending from LIKE to WANT to NEED and that the phenomena of these states and the transitions among them are explicable in terms of dopamine receptor and dopamine transporter regulation.
Abbreviations
DA: Dopamine; VTA: Ventral Tegmental Area; D1R: Dopamine D1-like Receptor; D2R: Dopamine D2-like Receptor; GABAR: GABA A Receptor; DAT: Dopamine Transporter; ACC: Nucleus Accumbens; LH: Lateral Hypothalamus; MFB: Medial Forebrain Bundle; GLUR: Glutamate Receptor; PFC: Prefrontal Cortex; Ant Cing: Anterior Cingulate Cortex; Dorsal Str: Dorsal Striatum; MOA: Mechanism of Action; MSN: Medium Spiny Neurons; ML: Mesolimbic; MC: Mesocortical; NSS: Nigrostriatal System
Introduction
Today the phrase “use disorder” has replaced both addiction and dependence in the current literature (eg. DSM-V) because the focus is on the continuum of severity [1]. However, the phrase still acknowledges the essential feature of attachment to a substance that is largely beyond volitional control1.
1Because of this and the significance of the NEED state (discussed below) we will use the term dependence in the present review.
Considerable evidence suggests that genetic polymorphisms in critical genes affect the risk for drug dependence (severe substance use disorder). In severe stimulant use disorder the drug is consumed in larger amounts than intended in spite of explicit knowledge of adverse effects, the social and interpersonal problems exacerbated by uncontrolled drug use and even a strong motivation to terminate drug use [2-4]. We and others have suggested that this is based on the development of a state of NEED transitioning from a state of WANT in the dependence process [5-13].
The 2013 UN World Drug Report stated that 230 million people (5% of the world population) had used drugs at least once, with 27 million of them (0.6% of the world population) being dependent on some drug [13,14]. In the US 40 million people or 12% of the population meet the criteria for substance abuse disorder [15,16].
Methods
We have recently provided an integrated theory of severe substance use disorder (dependence) that integrates Incentive Sensitization, Receptor Down-regulation, Psychomotor Stimulant, and Opponent Process theories into a framework [17,18]. The literature that helped to derive these theories was integrated with recent publications by these and other authors from searches done on PubMed using search criteria as follows: review article, free full text, published since 2000, and drug addiction or drug dependence.
Results
Overview
One of the difficulties in developing a comprehensive theory of drug dependence (severe substance use disorder) is that complex and possibly different brain mechanisms exist for the phases of development and maintenance of the dependence. Thus, most formulations have tended to focus on a single phase rather than the entire process. To this end, the concepts of LIKE, WANT and NEED have been developed to attempt to describe respectively, the positively reinforcing aspects of early and intermediate drug use, and a later process in which the individual can only attain a perception of “normalcy” in the presence of the drug. While the LIKE, WANT, and NEED concepts are vital to an understanding of the dependence process, the transition between LIKE and WANT and between WANT and NEED is equally important to discern how the disorder develops and is maintained.
The neural substrates for LIKE, WANT and NEED include the ML/MC DA and the NSS pathways. To some extent these substrates are considered common to the major theories of drug use disorders. The VTA is the origin of the DA neurons that project through the MFB, via the LH to limbic archi-, paleo- and neocortex. The ML pathway is the projection through the MFB to the nucleus accumbens (ventral striatum, ACC), amygdala and septum, whereas the MC pathway projects to frontal and cingulate cortex. Termination areas of these two pathways differ dramatically in the regulation of DA synthesis/release because of a differential expression of autoreceptors [19,20]. The NSS extends from the more laterally lying DA neurons in the midbrain substantia nigra to the corpus striatum. Thus, three major neuroanatomically distinct DA afferent pathways can be distinguished. All major theories of drug dependency associate these pathways with the different stages and effects of drug usage [17,18,21,22].
LIKE and LIKE to WANT: Our integrative theory extended the Robinson and Berridge’s incentive sensitization formulation by explicitly postulating that the ML projection to the ACC is the LIKE substrate [2,5,17,18,23-25]. We also postulated that the afferent MC DA projection plus the efferent glutamatergic projection from medial prefrontal cortex (mPFC) to dorsal and ventral striatum are the neuroanatomical substrates for the transition from LIKE to WANT. One key to understanding these transitions is that the regulation of DA synthesis and release is less tightly controlled within the MC portions of the limbic system than in the ML portion because of different densities of autoreceptors as noted above.
Data suggest that the transient release of DA in the ACC in particular may be critical to the initiation of drug use (the LIKE phase) [3,4,18,22,26]. ACC DA release is a function of activation of DA neurons in the ventral tegmentum and substantia nigra [18]. Such activation may become conditioned to environmental or contextual stimuli that signal reinforcement [17,18,26,27]. Some of the neural components of the LIKE circuit have been specified [8]. The GABA medium spiny neurons of the striatum receiving DA input project to other GABA neurons in the ventral tegmentum and substantia nigra that inhibit DA neurons projecting back to the corpus striatum. This constitutes a negative feedback loop that tightly controls DA release (Figure 1). Too little DA in the ACC or dorsal striatum allows the medium spiny neurons to fire, inhibiting the midbrain GABA neurons and disinhibiting the DA neurons. Too much DA inhibits the medium spiny neuron that then allows the GABA neurons to increase inhibition of the DA neurons. Together, this reduces DA release in the ACC (Figure 1).
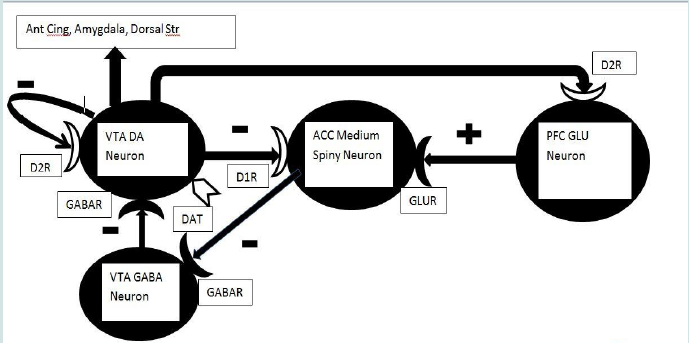
DA release from VTA DA neurons will inhibit medium spiny neurons in ACC. In turn, the medium spiny neurons will then fail to inhibit GABA neurons in the VTA, which will consequently inhibit VTA DA neuronal release of DA. This reduction of DA release will disinhibit the medium spiny neurons which will then inhibit VTA GABA neurons, disinhibiting the VTA DA neurons via this long feedback loop. A second feedback loop consists of DA inhibition of GLU neurons in mPFC which will reduce glutamatergic activation of medium spiny neurons again resulting in transient inhibition of DA release in VTA as above. Any elevation in DA release should be transient and opposed by D2-like autoceptors on DA neuron terminal endings (not shown), dendrites and somae (a short feedback loop), as well as the two long feedback loops. The transient elevation of DA release may in turn modulate neuronal activity in other mesocortical and mesolimbic projections sites (e.g. Ant Cing, septum, amygdala). The transient elevation of DA release may be the neurochemical correlate of euphoria (i.e. “LIKE”).
Down-regulation of presynaptic or postsynaptic DA receptors on the DA VTA neuron, medium spiny neuron or Glu neuron in mPFC will result in elevated DA release. Any reduction in the activity of DAT should yield a similar elevation of DA in the synaptic cleft. This elevation may be the neurochemical correlate of “WANT.” However, as down-regulation proceeds it is possible that the DA neurons will be largely depleted of releasable DA and any further application of psychostimulant drugs may result in diminishing returns, i.e. further down-regulation of DA receptors and associated depletion of DA stores in VTA neurons. White coloration indicates potential sites of DA receptor down-regulation or reduced DAT activity.
If a DA receptor antagonist is applied directly in the ACC, the rewarding effects of VTA stimulation and the locomotor effects of amphetamine are blocked. Such effects are not seen with microinjection of the antagonist into the medial prefrontal cortex (mPFC) [19]. These studies suggest that ACC-neuron DA receptors are critical to the LIKE response. But repeated stimulant drug use causes down-regulation of both presynaptic D2-like DA receptors and DAT, as is expected in a transition from LIKE to WANT.
WANT and WANT to NEED: A genetic polymorphism in DAT heightens drug cue-related responses in cocaine-dependent individuals in short-term cocaine withdrawal, a condition that certainly produces WANT [28]. In individuals lacking the DAT polymorphism, continued exposure to cocaine should likewise downregulate both presynaptic D2-like DA receptors as well as DAT and elevate DA release. This results in a sensitized response to DA. But as dependency develops postsynaptic DA receptors in ACC and PFC will down-regulate with a corresponding reduction in euphoria produced by the stimulant drug. Furthermore, an increased activity of the descending cortical glutamatergic projection to the ACC, due to down-regulation of postsynaptic mesocortical DA receptors, could produce the same result (WANT) as a down-regulation of D2-like DA autoreceptors on the mesolimbic projection to ACC [29].
Both behavioral sensitization and the associated heightened release of DA in the ACC can be largely blocked by the activation of postsynaptic D2 receptors in the medial prefrontal cortex (mPFC) [29]. Autoreceptors regulating DA release are differentially expressed in the forebrain DA projections. They are enriched within the corpus striatum, expressed moderately within the subcortical limbic system but are virtually absent from the frontal cortex broadly defined [21,30]. The specific projection to the PFC has DA autoreceptors that modulate release but not synthesis [20,30]. In addition, the dopaminergic projection to PFC lacks DAT (instead, DA is taken up by the norepinephrine transporter). Together these observations suggest that the MC projection is not under the tight negative feedback seen in the striatal projection and therefore DA release might be greatly potentiated by stimulant drugs, at least during the WANT phase. Ultimately, this could lead to the down-regulation of PFC D2 receptors and potentiation of the activity of the descending glutamatergic cortical projection to ACC. That potentiation, by stimulating the medium spiny neuron (which would then inhibit the tegmental GABA neuron) could further elevate DA release and consequent behavioral sensitization (WANT) (Figure 1). An exciting report indicates that a genetic mutation in the Cytoplasmic FMRP Interacting Protein 2 largely eliminates sensitization to cocaine and amphetamine in the mouse. However, at this time mechanistic details are lacking [31].
A reasonable hypothesis is that the WANT stage of the use disorder process for stimulant drugs in part reflects behavioral sensitization produced by a combination of the above mechanisms. Together, these actions on limbic structures result in increased DA release in the ACC and increased seeking for the drug. This is an example of positive feedback not subject to the normal negative feedback constraints (Figure 1). Mechanisms to increase DA release could include D2-like autoreceptor down-regulation, a functional change in DAT or a facilitation of the descending glutamatergic projection from mPFC to ACC. The mechanism must be powerful enough to overcome the negative feedback associated with DA inhibition of the medium spiny GABA neuron or DA inhibition of the firing of the mPFC glutamatergic projection neuron. Otherwise, the actions of these DA projections would ultimately restore inhibition of the DA neurons by local GABA neurons in the VTA. Stimulant drugs such as amphetamine or cocaine may accomplish this antagonism of negative feedback by a direct effect on the DA neuron as opposed to drugs that act through the medium spiny neuron and associated network (Figure 1).
Thus, WANT implies both behavioral sensitization and an orientation to those environmental stimuli that predict drug availability. We also postulate that WANT implies automaticity in the instrumental response that obtains the drug (for review see [32]). Significantly, we further suggest that euphoria may still be present in the WANT stage of severe drug use disorder development, although diminished when compared with that in the LIKE state.
We suggest that WANT explicitly leads to the transition from moderate substance use disorder (formerly abuse) to severe substance use disorder (dependence or NEED). Animal models often exhibit behavioral facilitation or sensitization as a measurable correlate of the WANT state [33-38]. Reductions in the density of axon terminal DA autoreceptors in the ACC or dorsal striatum as drug use accelerates could produce sensitization via a reduced inhibition of DA release [39]. In fact, the literature suggests that down-regulation of the D2- like DA autoreceptors could lead to elevated DA synthesis, release and metabolism in the ACC or other postsynaptic sites [8,10,11,39-43]. Changes in receptor density have been measured directly following treatments with agonists that induce behavioral sensitization. Under these conditions, the measures necessarily reflect both presynaptic and the much more numerous postsynaptic heteroreceptors. In this instance the results have not conclusively shown reductions in receptors [39]. We note that such changes in postsynaptic DA receptors would be most consonant with the NEED state, see below [12,44]. Changes in D2-like auto-receptors might better characterize the WANT state, but would be more difficult to observe because of a swamping effect due to the numerical dominance of postsynaptic DA receptors.
A reduction in the maximal transport capacity of the DAT, possibly mediated by an altered phosphorylation state could lead to elevated DA levels in the synaptic cleft and behavioral sensitization to the drug that could endure for as long as the drug continues to be used [45,46]. Thus, the DAT knockout mouse exhibits a 400% increase in locomotor behavior, as well as a 500% elevation in extracellular DA and a 300% increase in half-life [47,48]. Furthermore, the adaptations that have occurred in the expression of transport proteins, receptors and other key signalling molecules could induce craving in the absence of the drug, although craving seems more characteristic of NEED rather than WANT.
NEED: In the person with severe stimulant use disorder, withdrawal from the drug results in variable adverse effects, depending on a host of variables including the drug type, route of administration, frequency of use, dosage and genetic factors. It is important to say however that withdrawal from stimulant drugs is milder than withdrawal from most other drugs of abuse. However, craving can persist for many years after the individual has remained drug-free. This in turn is a major factor in drug recidivism. The persistence of craving implies and neurobiological research has confirmed that severe stimulant use disorder changes brain chemistry. Ultimately, these changes are the basis for NEED. It is reasonable to view the NEED state as one in which extinction of previously reinforcing behaviors occurs and is replaced by a desire to avoid the aversive consequence of withdrawal, even if those consequences are relatively mild [9-11,43-46,49]. However, this view is not necessarily totally compatible with the changes in brain chemistry that have been observed [17,18].
But if withdrawal symptoms are relatively mild and drug-related euphoria is greatly reduced why does foraging for drugs persist? We speculate that foraging for drugs is a behavior that is only occasionally successful, resulting in partial reinforcement of that behavior. Partially reinforced behaviors persist for a very long time in extinction, so this associative phenomenon may be one factor contributing to the persistence of drug foraging in the NEED state. Perhaps more importantly, when an abstinent drug user simply observes drug use paraphernalia that behaviour induces both craving and an activation of brain structures previously activated by drug ingestion [50,51]. Just as there is both an appetitive (motor activation) and a consummatory component (euphoria or pleasure or reinforcement) to dopaminergic activation, there appears to be both an appetitive (craving) and a consummatory (brain region activation) component in subjects exposed to these stimuli while in the NEED state [52,53]. It may be that exposure to such secondary reinforcers (i.e., drug paraphernalia) may activate regions downstream from the dopamine neuron but nevertheless components of the euphoric response to stimulant drugs of abuse. If such secondary reinforcers are internalized, simply thinking about drug paraphernalia may induce craving.
As the Receptor Down-Regulation theory suggests, NEED seems to occur in the absence or significantly reduced expression of euphoria [3,4,6,54]. This may be because DA stores have largely been depleted in the WANT stage through constant release or uptake inhibition and associated down-regulation of D2 receptors in the ACC or mPFC. Therefore, high rates of drug seeking are maintained, in part, in the attempt to avoid the dysphoria associated with excessive DA receptor down-regulation and potential DA depletion in the ACC, dorsal striatum and other DA projection sites. With psychostimulant drugs, the prior WANT-associated DA depletion may exacerbate the NEED state [17,18].
It has been difficult to demonstrate receptor down-regulation with stimulant drugs using rodent models. For instance striatal D2 receptor down-regulation has been observed with 1, 3 or 5 days of pretreatment with amphetamine or N-propylapomorphine (a direct DA agonist), but not with apomorphine (another direct DA agonist) [40]. However, PET studies in primates have consistently reported a decrease in D2 receptor number in the striatum with at least 14 days of treatment with amphetamine [55-57]. The rodent and primate models differed with respect to the mode of drug delivery and of course in the anatomy of the mesocortical system [42,55-57].
Fundamentally, WANT and NEED engage neural mechanisms that are associated with survival of the organism (air, food, water) or of the species (sex, social interactions for mammals, etc.). The consumption of food or water is pleasurable and that pleasure reinforces consumption. But stimulant drugs bypass behavior and produce euphoria by direct stimulation of the dopamine system. In fact, rodents who self-stimulate in the pleasure pathway of the MFB ay do that to the complete neglect of eating and drinking.
Our formulation explains the clinical observation that weak or partial direct DA agonists are more effective in preventing craving as opposed to a variety of agents than are competitive antagonists at the respective receptors [18]. This is because the latter precipitate the dysphoria associated with complete blockade of the relevant receptors, whereas the former provide mild tonic receptor stimulation. This idea is similar to the effects of receptor down-regulation in the classic Receptor Down-Regulation and Psychomotor Stimulant theories.
Conclusion
Based on substantial data, we hypothesize that substance use and abuse develops over three phases, LIKE, WANT and NEED. In each case drug use leads to predictable changes in DA presynaptic and postsynaptic receptors in ACC and mPFC. In LIKE, the initial response to psychostimulant drugs is highly reinforcing but limited by negative feedback. In WANT, as drug ingestion becomes chronic, negative feedback is “broken” by down-regulation of the DA transporter as well as DA receptors in ACC and mPFC, resulting in a hyper-dopaminergic condition and behavioral sensitization. This positive feedback cannot continue indefinitely as dopamine stores are depleted in NEED and euphoria is greatly reduced. But foraging for drugs persists because of craving, which may reflect exposure to secondary reinforcers such as drug paraphernalia in the user’s environment or even as internalized in the user’s thought process [3,4,8,58,59].
References
- National Institute on Alcohol Abuse and Alcoholism (2016) Alcohol use disorder: a comparison between DSM-IV and DSM-5.
- Robinson TE, Berridge KC (2008) Review. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc Lond B Biol Sci 363: 3137-3146.
- Volkow ND, Koob GF, McLellan AT (2016) Neurobiologic advances from the brain disease model of addiction. N Engl J Med 374: 363-371.
- Volkow ND, Wang GJ, Fowler JS, Tomasi D (2012) Addiction circuitry in the human brain. Annu Rev Pharmacol Toxicol 52: 321-336.
- Robinson TE, Berridge KC (2003) Addiction. Annu Rev Psychol 54: 25-53.
- Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F (2009) Imaging dopamine's role in drug abuse and addiction. Neuropharmacology 56 Suppl 1: 3-8.
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, et al. (2008) Dopamine increases in striatum do not elicit craving in cocaine abusers unless they are coupled with cocaine cues. Neuroimage 39: 1266-1273.
- Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94: 469-492.
- Koob GF, Caine SB, Parsons L, Markou A, Weiss F (1997) Opponent process model and psychostimulant addiction. Pharmacol Biochem Behav 57: 513-521.
- Koob GF, Le Moal M (1997) Drug abuse: hedonic homeostatic dysregulation. Science 278: 52-58.
- Koob GF, Le Moal M (2008) Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci 363: 3113-3123.
- Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35: 217-238.
- Wise RA, Koob GF (2014) The development and maintenance of drug addiction. Neuropsychopharmacology 39: 254-262.
- 14.United Nations Office on Drugs and Crime (2013) World Drug Report 2013. United Nations Publication. New York, USA.
- Sederer L (2015) A blind eye to addiction. U.S. News and World Report.
- Hedegaard H, Warner M, Minino AM (2017) Drug overdose death in the United States, 1999-2015. Centers for Disease Control and Prevention (CDC), NCHS Data Brief No. 273.
- Pakdaman S, Wilcox RE, Miller JD (2014) Theories and treatment of drug dependency: a neurochemical perspective. Curr Mol Pharmacol 7: 52-66.
- Wilcox R, Miller JD (2016) Molecular mechanisms of drug abuse, dependency and craving. J Addict Depend 2: 1-12.
- Wilcox R, Gonzales RA, Miller JD (1998) Introduction to neurotransmitters, receptors, signal transduction and second messengers. In: Nemeroff CB, Schatzberg AF (Eds), Textbook of Psychopharmacology.
- Wilcox R (1999) Dopamine. In: Craighead WE, Nemeroff CB (Eds), Encyclopedia of Psychology & Neuroscience.
- Anthony JC, Warner LA, Kessler RC (1994) Comparative epidemiology of dependence on tobacco, alcohol, controlled substances, and inhalants: basic findings from the National Comorbidity Survey. Exp Clin Psychopharmacol 2: 244-268.
- Wilcox RE, McMillen BA (1998) The rational use of drugs as therapeutic agents for the treatment of the alcoholisms. Alcohol 15: 161-177..
- Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18: 247-291.
- Robinson TE, Berridge KC (2000) The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95 Suppl 2: S91-S117.
- Robinson TE, Berridge KC (2001) Incentive-sensitization and addiction. Addiction 96: 103-114.
- Wilcox RE, Erickson CK (2011) The brain biology of drug abuse and addiction. In: McNeece CA, DiNitto DM (Eds), Chemical dependency: a systems approach. Pearson Education, Inc., USA, pp. 600.
- Moeller SJ, Parvaz MA, Shumay E, Beebe-Wang N, Konova AB, et al. (2013) Gene x abstinence effects on drug cue reactivity in addiction: multimodal evidence. J Neurosci 33: 10027-10036.
- Zadina JE, Chang SL, Ge LJ, Kastin AJ (1993) Mu opiate receptor down-regulation by morphine and up-regulation by naloxone in SH-SY5Y human neuroblastoma cells. J Pharmacol Exp Ther 265: 254-262.
- Jankelowitz SK (2013) Treatment of neurolept-induced tardive dyskinesia. Neuropsychiatr Dis Treat 9: 1371-1380.
- Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A 85: 5274-5278.
- Kumar V, Kim K, Joseph C, Kourrich S, Yoo SH, et al. (2013) C57BL/6N mutation in cytoplasmic FMR interacting protein 2 regulates cocaine response. Science 342: 1508-1512.
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, et al. (2013) Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341: 1521-1525.
- Riffee WH, Wilcox RE (1985) Effects of multiple pretreatment with apomorphine and amphetamine on amphetamine-induced locomotor activity and its inhibition by apomorphine. Psychopharmacology (Berl) 85: 97-101.
- Riffee WH, Wilcox RE (1987) Inhibition of amphetamine-induced locomotor activity by S-(+)-apomorphine: comparison with the action of R-(-)-apomorphine. J Pharm Pharmacol 39: 71-72.
- Wilcox RE, Riffee WH, Chen PC, Hammett S 3rd, Smith RV (1980) Behavioral facilitation following chronic administration of N-n-propylnorapomorphine. Psychopharmacology (Berl) 72: 113-115.
- Wilcox RE, Severson JA, Woodward JJ, Randall PK, Vaughn DM, et al. (1990) Behavioral sensitization following a single apomorphine pretreatment--selective effects on the dopamine release process. Brain Res 528: 109-113.
- Wilcox RE, Smith RV, Anderson JA, Riffee WH (1980) Apomorphine-induced stereotypic cage climbing in mice as a model for studying changes in dopamine receptor sensitivity. Pharmacol Biochem Behav 12: 29-33.
- Luo AH, Tahsili-Fahadan P, Wise RA, Lupica CR, Aston-Jones G (2011) Linking context with reward: a functional circuit from hippocampal CA3 to ventral tegmental area. Science 333: 353-357.
- Vaughn DM, Severson JA, Woodward JJ, Randall PK, Riffee WH, et al. (1990) Behavioral sensitization following subchronic apomorphine treatment--possible neurochemical basis. Brain Res 526: 37-44.
- Riffee WH, Wilcox RE, Vaughn DM, Smith RV (1982) Dopamine receptor sensitivity after chronic dopamine agonists. Striatal 3H-spiroperidol binding in mice after chronic administration of high doses of apomorphine, N-n-propylnorapomorphine and dextroamphetamine. Psychopharmacology (Berl) 77: 146-149.
- McEwen BS (2006) Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci 8: 367-381.
- McEwen BS, McKittrick CR, Tamashiro KL, Sakai RR (2015) The brain on stress: insight from studies using the visible burrow system. Physiol Behav 146: 47-56.
- Koob GF (2013) Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci 13: 3-30.
- Koob GF (2010) The role of CRF and CRF-related peptides in the dark side of addiction. Brain Res 1314: 3-14.
- Koob GF (2009) Brain stress systems in the amygdala and addiction. Brain Res 1293: 61-75.
- Koob GF (2009) Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56 Suppl 1: 18-31.
- Everitt BJ (2014) Neural and psychological mechanisms underlying compulsive drug seeking habits and drug memories--indications for novel treatments of addiction. Eur J Neurosci 40: 2163-2182.
- Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, et al. (2008) Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci 363: 3125-3135.
- Koob GF (2015) The dark side of emotion: the addiction perspective. Eur J Pharmacol 753: 73-87.
- Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, et al. (1999) Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156: 11-18.
- Kober H, Lacadie CM, Wexler BE, Malison RT, Sinha R, et al. (2016) Brain activity during cocaine craving and gambling urges: an fMRI study. Neuropsychopharmacology 41: 628-637.
- Miller JD, Sanghera MK, German DC (1981) Mesencephalic dopaminergic unit activity in the behaviorally conditioned rat. Life Sci 29: 1255-1263.
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, et al. (2001) Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry 58: 334-341.
- Volkow ND, Fowler JS, Wang GJ (2004) The addicted human brain viewed in the light of imaging studies: brain circuits and treatment strategies. Neuropharmacology 47 Suppl 1: 3-13.
- Enoksson T, Bertran-Gonzalez J, Christie MJ (2012) Nucleus accumbens D2- and D1-receptor expressing medium spiny neurons are selectively activated by morphine withdrawal and acute morphine, respectively. Neuropharmacology 62: 2463-2471.
- Ersche KD, Jones PS, Williams GB, Turton AJ, Robbins TW, et al. (2012) Abnormal brain structure implicated in stimulant drug addiction. Science 335: 601-604.
- Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, et al. (2010) Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 30: 17051-17062.
- Wise RA (2009) Roles for nigrostriatal--not just mesocorticolimbic--dopamine in reward and addiction. Trends Neurosci 32: 517-524.
- Wise RA, Morales M (2010) A ventral tegmental CRF-glutamate-dopamine interaction in addiction. Brain Res 1314: 38-43.