
Research Article
*Address for Correspondence: Bronwyn Kivell, School of Biological Sciences, Centre for Biodiscovery, Victoria University of Wellington, PO Box 600, Wellington 6140, New Zealand, Tel: (+64) 4 463 5233; Fax: (+64) 4 4635331; E-mail:Bronwyn.kivell@vuw.ac.nz
Citation: Holley A, Simonson B, Kivell BM. MDMA regulates serotonin transporter function via a Protein kinase C dependent mechanism. J Addiction Prevention. 2013;1(1): 5.
Copyright © 2013 Holley A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Addiction & Prevention |ISSN: 2330-2178 | Volume: 1, Issue: 1
Submission: 18 June 2013| Accepted: 15 July 2013 | Published: 17 July 2013
Modulation of SERT function is controlled through a multitude of signaling networks present in the cell. These networks are made up of pathways which interlink and regulate one another; some of these are constitutively in place, while others are activated upon extracellular signals. There are three major protein kinases, protein kinase C (PKC) [6], protein kinase A (PKA) [7] and P38-Mitogen activated protein kinase (P38-MAPK) [8] responsible for SERT’s phosphorylation state which dictates the rate of SERT insertion into the membrane, SERT function and SERT trafficking [3,9]. The specific residues responsible for regulation of SERT by molecules such as kinases are not completely understood. It appears that basal phosphorylation occurs at separate sites to those responsible for rapid regulation of SERT [7,10]. It is commonly known that SERT function is regulated by trafficking between the plasma membrane and intracellular compartments through a series of post-translational modifications [3, 7,10,11]. However, the mechanisms underlying how MDMA interacts and affects the signaling networks responsible for SERT regulation is unclear. Studies carried out in our laboratory have shown that MDMA is able to rapidly internalize SERT from the plasma membrane into the cytosol in HEK-293 and N2A cells thereby decreasing SERT cell surface localization and function [11]. What is not clear; however, is the mechanism by which this decrease in SERT function occurs. We have shown previously that it is independent of phospho-P38-MAP kinase.
PKC activation is correlated with an increased level of SERT phosphorylation which causes a reduction in both SERT function and cell surface expression [6,7]. Therefore, we investigated whether changes in SERT function caused by MDMA occurred via PKC-dependent signaling pathways. Being able to determine the biochemical pathways that are affected by MDMA in cell models will help to fill a gap in knowledge about how MDMA regulates SERT and may lead to the development of pharmacotherapies to prevent or reverse these changes.
Cells were grown in a Heracell incubator (Kendro Laboratory Products, GmbH, Germany) in a humid environment at 37ºC and supplemented with 5% CO2. All cell work was carried out in a class II biological safety cabinet (AES Environment Pty LTD, Auburn, Australia) with HEPE air filters. Human embryonic kidney cells (HEK-293) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in Dulbeccos modified Eagle medium (DMEM) (Gibco, Invitrogen, Auckland, NZ) containing 10% fetal calf serum (FCS) (ICP Biologicals, Auckland, NZ)), 1% Penstrep antibiotic (Penicillin G sodium 5000 units/ mL. Streptomycin sulphate 5000 units/mL in 0.85% saline, Gibco, Invitrogen; Auckland, New Zealand) and 200 μM L-glutamine (Gibco, Invitrogen). DMEM media containing no penstrep antibiotic (containing 200 μM L-glutamine and 10% FCS) was used during transfection. The selective PKC inhibitor Bisindolylmaleimide I (Bis I) (Sigma Aldrich, Castle Hill, NSW, Australia) dissolved in DMSO was added to the cell culture dishes at a final concentration of 200 nM. Bis I is a potent and selective PKC inhibitor previously shown to block activation of PKC by β-PMA in astrocytes [12]. Studies in both tissue and cell preparations have shown that pre-treatment with Bis I for 30-90 min is sufficient to inhibit PKC [13,14]. PKC activator β-phorbol 12 myristate 13-acetate (β-PMA) (1 μM) (Sigma Aldrich), a known regulator of SERT was used as described previously [8].
On day 1 HEK-293 cells were plated into 100 mm culture dishes (Becton Dickinson Ltd, Auckland, New Zealand) in 7 mL media at density of 1x106 cells/mL, on day 2, when growth reached 60-70% confluency, cells were transiently transfected with the enhanced green fluorescent protein human tagged serotonin transporter (EGFP-hSERT) plasmid (0.5 μg/mL) (kindly donated by Professor Sammanda Ramamoorthy; Virginia Commonwealth University, Richmond, VA) using Lipofectamine 2000 reagent (Invitrogen) following the manufactures protocol. Dishes containing the cells to be transfected had old culture medium replaced with fresh penstrepfree medium and cells used 48 hours after transfection.
Rotating Disc Electrode Voltammetry (RDEV)
RDEV is an electrochemical technique that has been previously used for the measurement of monoamine transporter function in real time [15,16]. Uptake of 5-HT in tissue samples such as whole brain rat synaptosomes has also been described recently [17]. Here, RDEV is used to investigate the changes in SERT function as defined by rate of change of 5-HT uptake under different treatments in HEK-293 transiently expressing SERT. A rotating glassy carbon electrode (Pine Instruments, special order AFMDO3GC, Pennsylvania) connected to a high-precision MSR rotator (Pine Instruments) was rotated at 2000 rpm. An Ag/AgCl reference electrode was used to apply a +450 mV potential relative to the electrode by an eDAQ potentiostat EA161 (eDAQ, NSW, Australia). Chart software (eDAQ, NSW, Australia) was used to save the resulting currents recorded by the eDAQ recorder 210 (eDAQ, NSW, Australia).
Preparation of EGFP-Hsert Transfected HEK-293 for RDEV
Cells transiently transfected with EGFP-hSERT were treated with compounds so that their maximal effect on SERT was achieved, then washed 5 x times with 37ºC KREBs buffer saturated with 5%CO2/95%O2. They were harvested by mechanical agitation with the KREBs buffer and centrifuged at 300 g for 5 min. The pellet of cells were then resuspended in 1.2 mL of KREBs buffer saturated with carbogen (5%CO2/95%O2) and maintained at 37ºC for up to 20 min until needed. For 5-HT uptake experiments, 300 μL of EGFPhSERT expressing cells were added to the glass chamber and the rotator lowered into the center and rotated at 2000 rpm until a stable baseline recording was achieved. A single addition of 5-HT (1.5 μM) was added to the cell suspension, and uptake recorded for 20s. A 10 μL aliquot of cell suspension containing 0.4% tryphan blue was used to determine cell number using a haemacytometer. Transfection percentages were calculated by visualization of EGFP-hSERT positive cells using a fluorescent microscope (AX70, Olympus, Mt Waverly, Australia).
Data analysis
SERT uptake was determined from a 10 s period directly after addition of 5-HT. Microsoft Office Excel was used to plot the data and generate a linear regression. A linear regression with an R2 value less than 0.95 was discarded from this study. The change in current recorded from measurement period of 5-HT uptake by the cells was transformed into μmol/s using a standard concentration curve generated at the beginning of the experiment. Using data generated from the standard curve and cell numbers, SERT uptake was expressed in pmol/s/106 cells. The values were then entered into GraphPad Prism Version 5.04 for Windows, (GraphPad Software Inc., San Diego California USA) to calculate the kinetic data. Oneway ANOVA and Student t-tests were used where appropriate to determine the statistical significance.
MDMA is known to compete with 5-HT for uptake by SERT [2]. We performed control experiments where MDMA (52 μM) was added to a 35 mm petri dish containing EGFP-hSERT transfected cells for 30 s and then removed and cells washed. 5-HT uptake was then measured using RDEV with a single addition of 5-HT at 1.5 μM. No change in 5-HT uptake was observed compared to control indicating that MDMA is able to be washed out and that a 30 sec MDMA exposure is not sufficient to cause trafficking related changes in SERT function (data not shown). To determine whether MDMA causes a down-regulation of SERT function transiently EGFP-hSERT expressing HEK-293 were incubated with 52 μM of MDMA for 5, 10, 30, 60 and 90 min prior to removal and washing. Cells incubated with MDMA for 5 min show that 5-HT uptake is significantly decreased compared to controls (Figure1), with MDMA reducing uptake by 47% (p<0.01). After a 10 min incubation with MDMA, uptake was reduced by 63% (p<0.001). This decrease in 5-HT uptake persisted until the 90 min time point (p<0.01). This demonstrates that MDMA caused a functional down-regulation of SERT that persisted for at least 90 min.
Effect of PKC Activator β-PMA on SERT Function
Once we established that MDMA caused a functional downregulation of SERT through RDEV methods (Figure1), the next step was to investigate whether it was caused by activation of PKC. Inhibition of SERT function occurs upon PKC activation, causing a reduction in 5-HT uptake (indicated by a decrease in Vmax) with little or no change in the affinity of SERT for 5-HT (no change in Km) [6]. Addition of 1 &mu:M β-PMA has been previously shown to decrease SERT function in HEK-293 cells within 20-60 min [6,11]. In this study, HEK-293 cells transiently transfected with EGFP-hSERT were pretreated with 1 &mu:M β-PMA for 30 or 60 min, followed by analysis of 5-HT uptake by RDEV. In HEK-293 cells, the 30 min pre-treatment with β-PMA significantly decreased uptake by 35% compared to controls (p<0.05) and treatment for 60 min reduced uptake by 45% (p<0.01) (Figure 2).
Effect of MDMA on 5-HT Uptake after PKC Inhibition
Results presented in Figures 1 and 2 shows that both MDMA treatment and PKC activation by β-PMA significantly decreased 5-HT uptake by SERT in HEK-293 cells. To establish whether MDMA modulates SERT down-regulation via a PKC dependent mechanism, cells were pre-treated with PKC inhibitor Bis I. HEK-293 cells transiently transfected with EGFP-hSERT were pretreated with 200 nM Bis I for 30 min. MDMA (52 &mu:M ) was then added for 5 min and 5-HT uptake measured. Pre-treatment with Bis I showed that 5-HT uptake was not reduced following MDMA treatment (Figure 3). Additionally, cells treated with vehicle show a significant decrease in 5-HT uptake following MDMA treatment (Figure 3). This shows that PKC inhibition prevents the effects MDMA has on decreasing SERT function.
Utilizing RDEV methods and cells expressing EGFP-hSERT we investigated the time course effects of MDMA on SERT function. MDMA was incubated for up to 90 min with serotonin 5-HT uptake measured at 5, 10, 30, 60 and 90 min. It was expected that MDMA would cause a rapid and sustained decrease in 5-HT uptake, since previous studies have shown that SERT is internalized after MDMA treatment [11]. Indeed, HEK-293 cells transfected with EGFP-hSERT displayed a significant decrease in 5-HT uptake (47-63%) following MDMA treatment in a rapid (within 5 min) and sustained (up to 90 min) manner ( Figure1). HEK-293 cells show a sustained decrease in SERT function after MDMA treatment that persisted for at least 90 min. These effects were not due to MDMA competing with 5-HT for uptake as MDMA was shown to be washed out prior to 5-HT additions (data not shown).
Previous studies have attempted to identify the pathways responsible for MDMA-induced regulation of SERT. Previously, P38-MAPK, a known regulator of SERT membrane expression [8,24,25] was not activated in response to MDMA treatment in EGFPhSER expressing HEK-293 and N2A cells, suggesting a P38-MAPK independent mechanism [11]. This led us to undertake the current study. We chose to investigate PKC, as it has been shown to be a regulator of SERT function. Activation of PKC by β-PMA leads to a decrease in SERT activity [26,27]. Previous studies also showed that MDMA caused an increase in PKC activity [28,29]. Here we show that treatment with PKC activator β-PMA for 30 or 60 min causesa decrease in 5-HT uptake in HEK-293 cells transfected with EGFPhSERT (Figure3). This finding provides evidence for a mechanism of how MDMA exerts its effects on SERT. By understanding the mechanism underlying these effects, better therapeutic targets can be developed to either reverse the long-term effects associated with MDMA use, or by preventing these neurochemical changes from occurring with MDMA use. It does not rule out additional signaling mechanisms that may also contribute to MDMA regulation of SERT, such as PKA [7], protein kinase G (PKG) [30], or protein phosphatase 1/2A [31] -dependent signaling pathways which will need to be investigated further. While there is no doubt that SERT is the primary regulator for 5-HT uptake, it must be noted that alternative mechanisms do play a role. Transporter types such as the organic cation transporters (OCTs) [32-34] or the plasma membrane monoamine transporter (PMAT) [35,36] have been shown to be capable of 5-HT uptake, and can come into play when 5-HT levels are high or when SERT function or expression is impaired or absent [33].
A related study has investigated the effects of amphetamine on the dopamine transporter (DAT) and has been shown to be regulated in a PKC-independent pathway following amphetamine-mediated down-regulation [37]. Even though amphetamine-stimulated dopamine efflux has been previously shown to be modulated through a PKC pathway [38,39], it appears that the amphetaminemediated sequestration of DAT is independent to this mechanism. It is not surprising that different monoamine transporters are regulated differently with related drugs of abuse, evidence for this is demonstrated widely within the literature [9]. This suggests that serotonin and dopamine transporters may be differentially regulated by amphetamine and MDMA. It is not unusual for different amphetamine drugs to target different pathways [10].
Bis I is a potent and selective PKC inhibitor, and studies have shown that it blocks the activation of PKC by β-PMA in astrocytes [12]. Previous studies in tissue and cell preparations have also shown that pre-treatment with Bis I for 30-90 min [13,14] is sufficient to inhibit PKC. In this study RDEV was used to measure any changes in 5-HT uptake in cells treated with MDMA in the presence or absence of Bis I. In this study, pre-treatment with Bis I blocked the decrease in 5-HT uptake caused by MDMA (Figure 3). These results conclude that MDMA regulates SERT function in this cellular model through a PKC dependent signaling pathway. Further investigations utilizing brain tissue and in vivo models will also expand on these findings to fully evaluate the role of PKC in modulating the effects of MDMA and the role this plays in modulating the neuroadaptations and neurodegeneration seen with MDMA use.
SERT regulation is tightly controlled by a large network of kinases which cross-talk with each other, creating an efficient 5-HT signaling system. How MDMA interacts and exerts its effects on the signaling molecules responsible for the regulation of SERT at the cell membrane would help to narrow the gap in scientists’ current knowledge about how drugs of abuse act on the monoamine systems. The neurotoxic action of MDMA is highly disputed throughout the literature, and although this subject was not a key focus in this study, advances in information about how MDMA targets SERT for downregulation may provide a stepping stone for further investigation into its mechanism of action. This is important for understanding how drugs of abuse are able to modulate their targets and this could lead to pharmacotherapies for reversing the effects of drugs of abuse or preventing drug-induced cellular changes.
MDMA Regulates Serotonin Transporter Function via a Protein Kinase C Dependent Mechanism
Holley A, Simonson B, and Kivell BM*
- School of Biological Sciences, Centre for Biodiscovery Victoria University of Wellington, Kelburn Parade, Wellington 6140, New Zealand
*Address for Correspondence: Bronwyn Kivell, School of Biological Sciences, Centre for Biodiscovery, Victoria University of Wellington, PO Box 600, Wellington 6140, New Zealand, Tel: (+64) 4 463 5233; Fax: (+64) 4 4635331; E-mail:Bronwyn.kivell@vuw.ac.nz
Citation: Holley A, Simonson B, Kivell BM. MDMA regulates serotonin transporter function via a Protein kinase C dependent mechanism. J Addiction Prevention. 2013;1(1): 5.
Copyright © 2013 Holley A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Addiction & Prevention |ISSN: 2330-2178 | Volume: 1, Issue: 1
Submission: 18 June 2013| Accepted: 15 July 2013 | Published: 17 July 2013
Abstract
Serotonin (5-HT) is a neurotransmitter with an integral role in regulating mood and dysregulation of this system is implicated in disorders such as depression and withdrawal from drugs of abuse. 3,4- methylenedioxymethamphetamine (MDMA) or ‘Ecstasy’ is a commonly abused drug which primarily targets the serotonin transporter (SERT) and competes with serotonin (5-HT) for uptake into the pre-synaptic neuron. By understanding how MDMA regulates SERT function it may be possible to target this system to prevent or reverse the changes seen with MDMA use. Previous studies have shown that MDMA is able to down-regulate SERT expression from the cell surface to intracellular vesicles, thereby decreasing 5-HT transport. How MDMA targets this regulatory pathway is unclear. Protein Kinase C (PKC) is a well-known regulator of SERT, and activation of PKC causes phosphorylation of SERT, targeting the transporter for internalization. In this study, using rotating disc electrode voltammetry (RDEV) we show that MDMA causes a significant decrease in SERT function in HEK-293 cells transiently expressing EGFP-hSERT. This MDMA-induced down-regulation was not observed when cells were pretreated with PKC inhibitor, Bis I. This data shows that the MDMA-induced downregulation of SERT occurs via PKC dependent signaling pathways.Keywords
Ecstasy; SERT; Transport; Regulation; Addiction; Drug abuseAbbreviations
MDMA: 3,4- methylenedioxymethamphetamine; SERT: Serotonin Transporter; PKC: Protein Kinase C; RDEV: Rotating Disc Electrode Voltammetry; Bis I: Bisindolylmaleimide I; 5-HT: Serotonin (5-Hydroxytryptophan); EGFP: Enhanced Green Fluorescent Protein; HEK-293: Human Embryonic Kidney-293; N2A: Mouse Neuroblastoma; DAT: Dopamine Transporter; NET: Norepinephrine transporter; DMEM: Dulbeccos modified Eagle mediumIntroduction
3,4-methylenedioxymethamphetamine (MDMA), commonly called ‘ecstasy’ is a ring-substituted amphetamine derivative. A drug of abuse popular among young people, it is a stimulant which induces hallucinogenic effects on users with feelings of euphoria and a heightened perception [1-3]. Short-term effects of MDMA include mild depression, hyperthermia, cardiac arrhythmia, hypertension, and memory loss. Prolonged or chronic use of MDMA is associated with long-term deficits in serotonin (5-HT) levels and loss of 5-HT nerve cell integrity [1-4]. Amphetamines have a high affinity for the monoamine transporters. MDMA exhibits the highest affinity for the SERT protein, with affinities for the dopamine (DAT) and norepinephrine (NET) transporter being tenfold less [2]. MDMA binds to SERT, competing for uptake with 5-HT into the nerve terminal. Once inside the cell, it reverses SERT transport and inhibits uptake of 5-HT from the synaptic cleft, thereby causing an initial increase in the extracellular pool of 5-HT. The cumulative effects of MDMA is that it causes a rapid efflux of 5-HT into the synaptic cleft leading to long-term depletion of 5-HT [2,5].Modulation of SERT function is controlled through a multitude of signaling networks present in the cell. These networks are made up of pathways which interlink and regulate one another; some of these are constitutively in place, while others are activated upon extracellular signals. There are three major protein kinases, protein kinase C (PKC) [6], protein kinase A (PKA) [7] and P38-Mitogen activated protein kinase (P38-MAPK) [8] responsible for SERT’s phosphorylation state which dictates the rate of SERT insertion into the membrane, SERT function and SERT trafficking [3,9]. The specific residues responsible for regulation of SERT by molecules such as kinases are not completely understood. It appears that basal phosphorylation occurs at separate sites to those responsible for rapid regulation of SERT [7,10]. It is commonly known that SERT function is regulated by trafficking between the plasma membrane and intracellular compartments through a series of post-translational modifications [3, 7,10,11]. However, the mechanisms underlying how MDMA interacts and affects the signaling networks responsible for SERT regulation is unclear. Studies carried out in our laboratory have shown that MDMA is able to rapidly internalize SERT from the plasma membrane into the cytosol in HEK-293 and N2A cells thereby decreasing SERT cell surface localization and function [11]. What is not clear; however, is the mechanism by which this decrease in SERT function occurs. We have shown previously that it is independent of phospho-P38-MAP kinase.
PKC activation is correlated with an increased level of SERT phosphorylation which causes a reduction in both SERT function and cell surface expression [6,7]. Therefore, we investigated whether changes in SERT function caused by MDMA occurred via PKC-dependent signaling pathways. Being able to determine the biochemical pathways that are affected by MDMA in cell models will help to fill a gap in knowledge about how MDMA regulates SERT and may lead to the development of pharmacotherapies to prevent or reverse these changes.
Materials and Methods
Tissue cultureCells were grown in a Heracell incubator (Kendro Laboratory Products, GmbH, Germany) in a humid environment at 37ºC and supplemented with 5% CO2. All cell work was carried out in a class II biological safety cabinet (AES Environment Pty LTD, Auburn, Australia) with HEPE air filters. Human embryonic kidney cells (HEK-293) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in Dulbeccos modified Eagle medium (DMEM) (Gibco, Invitrogen, Auckland, NZ) containing 10% fetal calf serum (FCS) (ICP Biologicals, Auckland, NZ)), 1% Penstrep antibiotic (Penicillin G sodium 5000 units/ mL. Streptomycin sulphate 5000 units/mL in 0.85% saline, Gibco, Invitrogen; Auckland, New Zealand) and 200 μM L-glutamine (Gibco, Invitrogen). DMEM media containing no penstrep antibiotic (containing 200 μM L-glutamine and 10% FCS) was used during transfection. The selective PKC inhibitor Bisindolylmaleimide I (Bis I) (Sigma Aldrich, Castle Hill, NSW, Australia) dissolved in DMSO was added to the cell culture dishes at a final concentration of 200 nM. Bis I is a potent and selective PKC inhibitor previously shown to block activation of PKC by β-PMA in astrocytes [12]. Studies in both tissue and cell preparations have shown that pre-treatment with Bis I for 30-90 min is sufficient to inhibit PKC [13,14]. PKC activator β-phorbol 12 myristate 13-acetate (β-PMA) (1 μM) (Sigma Aldrich), a known regulator of SERT was used as described previously [8].
On day 1 HEK-293 cells were plated into 100 mm culture dishes (Becton Dickinson Ltd, Auckland, New Zealand) in 7 mL media at density of 1x106 cells/mL, on day 2, when growth reached 60-70% confluency, cells were transiently transfected with the enhanced green fluorescent protein human tagged serotonin transporter (EGFP-hSERT) plasmid (0.5 μg/mL) (kindly donated by Professor Sammanda Ramamoorthy; Virginia Commonwealth University, Richmond, VA) using Lipofectamine 2000 reagent (Invitrogen) following the manufactures protocol. Dishes containing the cells to be transfected had old culture medium replaced with fresh penstrepfree medium and cells used 48 hours after transfection.
Rotating Disc Electrode Voltammetry (RDEV)
RDEV is an electrochemical technique that has been previously used for the measurement of monoamine transporter function in real time [15,16]. Uptake of 5-HT in tissue samples such as whole brain rat synaptosomes has also been described recently [17]. Here, RDEV is used to investigate the changes in SERT function as defined by rate of change of 5-HT uptake under different treatments in HEK-293 transiently expressing SERT. A rotating glassy carbon electrode (Pine Instruments, special order AFMDO3GC, Pennsylvania) connected to a high-precision MSR rotator (Pine Instruments) was rotated at 2000 rpm. An Ag/AgCl reference electrode was used to apply a +450 mV potential relative to the electrode by an eDAQ potentiostat EA161 (eDAQ, NSW, Australia). Chart software (eDAQ, NSW, Australia) was used to save the resulting currents recorded by the eDAQ recorder 210 (eDAQ, NSW, Australia).
Preparation of EGFP-Hsert Transfected HEK-293 for RDEV
Cells transiently transfected with EGFP-hSERT were treated with compounds so that their maximal effect on SERT was achieved, then washed 5 x times with 37ºC KREBs buffer saturated with 5%CO2/95%O2. They were harvested by mechanical agitation with the KREBs buffer and centrifuged at 300 g for 5 min. The pellet of cells were then resuspended in 1.2 mL of KREBs buffer saturated with carbogen (5%CO2/95%O2) and maintained at 37ºC for up to 20 min until needed. For 5-HT uptake experiments, 300 μL of EGFPhSERT expressing cells were added to the glass chamber and the rotator lowered into the center and rotated at 2000 rpm until a stable baseline recording was achieved. A single addition of 5-HT (1.5 μM) was added to the cell suspension, and uptake recorded for 20s. A 10 μL aliquot of cell suspension containing 0.4% tryphan blue was used to determine cell number using a haemacytometer. Transfection percentages were calculated by visualization of EGFP-hSERT positive cells using a fluorescent microscope (AX70, Olympus, Mt Waverly, Australia).
Data analysis
SERT uptake was determined from a 10 s period directly after addition of 5-HT. Microsoft Office Excel was used to plot the data and generate a linear regression. A linear regression with an R2 value less than 0.95 was discarded from this study. The change in current recorded from measurement period of 5-HT uptake by the cells was transformed into μmol/s using a standard concentration curve generated at the beginning of the experiment. Using data generated from the standard curve and cell numbers, SERT uptake was expressed in pmol/s/106 cells. The values were then entered into GraphPad Prism Version 5.04 for Windows, (GraphPad Software Inc., San Diego California USA) to calculate the kinetic data. Oneway ANOVA and Student t-tests were used where appropriate to determine the statistical significance.
Results
Effect of MDMA on SERT FunctionMDMA is known to compete with 5-HT for uptake by SERT [2]. We performed control experiments where MDMA (52 μM) was added to a 35 mm petri dish containing EGFP-hSERT transfected cells for 30 s and then removed and cells washed. 5-HT uptake was then measured using RDEV with a single addition of 5-HT at 1.5 μM. No change in 5-HT uptake was observed compared to control indicating that MDMA is able to be washed out and that a 30 sec MDMA exposure is not sufficient to cause trafficking related changes in SERT function (data not shown). To determine whether MDMA causes a down-regulation of SERT function transiently EGFP-hSERT expressing HEK-293 were incubated with 52 μM of MDMA for 5, 10, 30, 60 and 90 min prior to removal and washing. Cells incubated with MDMA for 5 min show that 5-HT uptake is significantly decreased compared to controls (Figure1), with MDMA reducing uptake by 47% (p<0.01). After a 10 min incubation with MDMA, uptake was reduced by 63% (p<0.001). This decrease in 5-HT uptake persisted until the 90 min time point (p<0.01). This demonstrates that MDMA caused a functional down-regulation of SERT that persisted for at least 90 min.
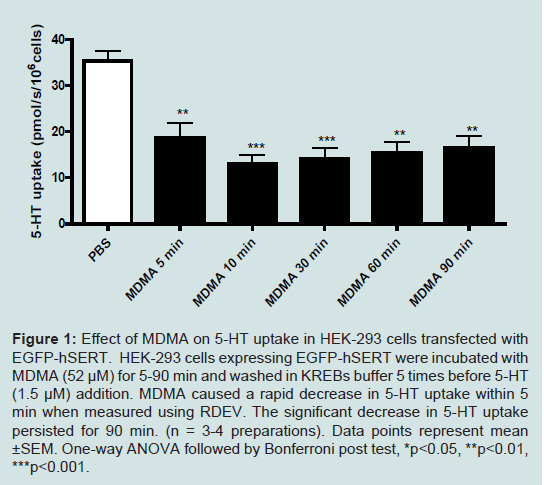
Figure 1: Effect of MDMA on 5-HT uptake in HEK-293 cells transfected with EGFP-hSERT. HEK-293 cells expressing EGFP-hSERT were incubated with MDMA (52 μM) for 5-90 min and washed in KREBs buffer 5 times before 5-HT (1.5 μM) addition. MDMA caused a rapid decrease in 5-HT uptake within 5 min when measured using RDEV. The significant decrease in 5-HT uptake persisted for 90 min. (n = 3-4 preparations). Data points represent mean ±SEM. One-way ANOVA followed by Bonferroni post test, *p<0.05, **p<0.01, ***p<0.001.
Once we established that MDMA caused a functional downregulation of SERT through RDEV methods (Figure1), the next step was to investigate whether it was caused by activation of PKC. Inhibition of SERT function occurs upon PKC activation, causing a reduction in 5-HT uptake (indicated by a decrease in Vmax) with little or no change in the affinity of SERT for 5-HT (no change in Km) [6]. Addition of 1 &mu:M β-PMA has been previously shown to decrease SERT function in HEK-293 cells within 20-60 min [6,11]. In this study, HEK-293 cells transiently transfected with EGFP-hSERT were pretreated with 1 &mu:M β-PMA for 30 or 60 min, followed by analysis of 5-HT uptake by RDEV. In HEK-293 cells, the 30 min pre-treatment with β-PMA significantly decreased uptake by 35% compared to controls (p<0.05) and treatment for 60 min reduced uptake by 45% (p<0.01) (Figure 2).
Figure 2: The effect of the PKC activator β-PMA on 5-HT uptake in HEK-293 cells transfected with EGFP-hSERT. HEK-293 cells were incubated with 1 μM of β-PMA for 30 or 60 min before 5-HT addition (1.5μM). A significant decrease in 5-HT uptake was seen in HEK-293 cells after 30 and 60 min β-PMA incubation (n = 4 preparations). Data points represent mean ±SEM. Student t-test; *p<0.05, **p<0.01, ***p<0.001.
Results presented in Figures 1 and 2 shows that both MDMA treatment and PKC activation by β-PMA significantly decreased 5-HT uptake by SERT in HEK-293 cells. To establish whether MDMA modulates SERT down-regulation via a PKC dependent mechanism, cells were pre-treated with PKC inhibitor Bis I. HEK-293 cells transiently transfected with EGFP-hSERT were pretreated with 200 nM Bis I for 30 min. MDMA (52 &mu:M ) was then added for 5 min and 5-HT uptake measured. Pre-treatment with Bis I showed that 5-HT uptake was not reduced following MDMA treatment (Figure 3). Additionally, cells treated with vehicle show a significant decrease in 5-HT uptake following MDMA treatment (Figure 3). This shows that PKC inhibition prevents the effects MDMA has on decreasing SERT function.
Figure 3: Effect of MDMA on 5-HT uptake after PKC inhibition by Bis I in HEK-293 cells transfected with EGFP-hSERT. HEK-293 cells transfected with EGFP-hSERT were incubated with either 200 nM of Bis I for 30 min, or incubated with 200 nM of Bis I for 30 min followed by incubation with 52 μM MDMA for 5 min before determining SERT uptake using RDEV with single 5-HT additions (1.5 μM). 5-HT uptake levels were compared to vehicle-treated controls (vehicle 1 is DMSO, vehicle 2 is PBS). Transfected cells were also incubated with 52 μM of MDMA for 5 min before, washout and 5-HT addition (1.5 μM). Data shows that pre-treatment with Bis I prevented the decrease in 5-HT uptake that MDMA causes, with 5-HT uptake levels comparable to vehicle treated control levels. (n = 3-5 for HEK-293 cell preparations). Data points represent the mean ± SEM. One-way ANOVA followed by Bonferroni post test, **p<0.01.
Discussion
The principal aim of this experiment was to identify the cellular signaling pathway responsible for the decrease in SERT function following exposure to MDMA. Traditional methods for studying SERT function in the brain include radioactive 5-HT uptake [7] high-speed chronoamperometry (HSC) [18] and fast scan cyclic voltammetry (FSCV) [19,20]. These techniques have been used to measure SERT function in cells, synaptosomes and in vivo tissue systems. Another technique used to measure neurotransmitter transporter function is called rotating disc electrode voltammetry (RDEV) [15]. This electrochemical technique is widely used in studies investigating the release and uptake of norepinephrine and dopamine by the neuronal monoamine transporters in both cell and brain tissue models [15,21,22]. Recently, RDEV has been validated for the measurement of SERT function in synaptosomes [17,23]. Cell models have been utilized throughout this study as they allow for greater manipulation when studying the effects of drugs on monoamine transporters than is possible in a whole animal or tissue samples. HEK-293 cells are a well-established cell model used to investigate monoamine transporter regulation and have been shown to contain many of the relevant cell signaling pathways.Utilizing RDEV methods and cells expressing EGFP-hSERT we investigated the time course effects of MDMA on SERT function. MDMA was incubated for up to 90 min with serotonin 5-HT uptake measured at 5, 10, 30, 60 and 90 min. It was expected that MDMA would cause a rapid and sustained decrease in 5-HT uptake, since previous studies have shown that SERT is internalized after MDMA treatment [11]. Indeed, HEK-293 cells transfected with EGFP-hSERT displayed a significant decrease in 5-HT uptake (47-63%) following MDMA treatment in a rapid (within 5 min) and sustained (up to 90 min) manner ( Figure1). HEK-293 cells show a sustained decrease in SERT function after MDMA treatment that persisted for at least 90 min. These effects were not due to MDMA competing with 5-HT for uptake as MDMA was shown to be washed out prior to 5-HT additions (data not shown).
Previous studies have attempted to identify the pathways responsible for MDMA-induced regulation of SERT. Previously, P38-MAPK, a known regulator of SERT membrane expression [8,24,25] was not activated in response to MDMA treatment in EGFPhSER expressing HEK-293 and N2A cells, suggesting a P38-MAPK independent mechanism [11]. This led us to undertake the current study. We chose to investigate PKC, as it has been shown to be a regulator of SERT function. Activation of PKC by β-PMA leads to a decrease in SERT activity [26,27]. Previous studies also showed that MDMA caused an increase in PKC activity [28,29]. Here we show that treatment with PKC activator β-PMA for 30 or 60 min causesa decrease in 5-HT uptake in HEK-293 cells transfected with EGFPhSERT (Figure3). This finding provides evidence for a mechanism of how MDMA exerts its effects on SERT. By understanding the mechanism underlying these effects, better therapeutic targets can be developed to either reverse the long-term effects associated with MDMA use, or by preventing these neurochemical changes from occurring with MDMA use. It does not rule out additional signaling mechanisms that may also contribute to MDMA regulation of SERT, such as PKA [7], protein kinase G (PKG) [30], or protein phosphatase 1/2A [31] -dependent signaling pathways which will need to be investigated further. While there is no doubt that SERT is the primary regulator for 5-HT uptake, it must be noted that alternative mechanisms do play a role. Transporter types such as the organic cation transporters (OCTs) [32-34] or the plasma membrane monoamine transporter (PMAT) [35,36] have been shown to be capable of 5-HT uptake, and can come into play when 5-HT levels are high or when SERT function or expression is impaired or absent [33].
A related study has investigated the effects of amphetamine on the dopamine transporter (DAT) and has been shown to be regulated in a PKC-independent pathway following amphetamine-mediated down-regulation [37]. Even though amphetamine-stimulated dopamine efflux has been previously shown to be modulated through a PKC pathway [38,39], it appears that the amphetaminemediated sequestration of DAT is independent to this mechanism. It is not surprising that different monoamine transporters are regulated differently with related drugs of abuse, evidence for this is demonstrated widely within the literature [9]. This suggests that serotonin and dopamine transporters may be differentially regulated by amphetamine and MDMA. It is not unusual for different amphetamine drugs to target different pathways [10].
Bis I is a potent and selective PKC inhibitor, and studies have shown that it blocks the activation of PKC by β-PMA in astrocytes [12]. Previous studies in tissue and cell preparations have also shown that pre-treatment with Bis I for 30-90 min [13,14] is sufficient to inhibit PKC. In this study RDEV was used to measure any changes in 5-HT uptake in cells treated with MDMA in the presence or absence of Bis I. In this study, pre-treatment with Bis I blocked the decrease in 5-HT uptake caused by MDMA (Figure 3). These results conclude that MDMA regulates SERT function in this cellular model through a PKC dependent signaling pathway. Further investigations utilizing brain tissue and in vivo models will also expand on these findings to fully evaluate the role of PKC in modulating the effects of MDMA and the role this plays in modulating the neuroadaptations and neurodegeneration seen with MDMA use.
SERT regulation is tightly controlled by a large network of kinases which cross-talk with each other, creating an efficient 5-HT signaling system. How MDMA interacts and exerts its effects on the signaling molecules responsible for the regulation of SERT at the cell membrane would help to narrow the gap in scientists’ current knowledge about how drugs of abuse act on the monoamine systems. The neurotoxic action of MDMA is highly disputed throughout the literature, and although this subject was not a key focus in this study, advances in information about how MDMA targets SERT for downregulation may provide a stepping stone for further investigation into its mechanism of action. This is important for understanding how drugs of abuse are able to modulate their targets and this could lead to pharmacotherapies for reversing the effects of drugs of abuse or preventing drug-induced cellular changes.
Acknowledgments
We would like to thank Professor Sammanda Ramamoorthy (Virginia Commonwealth University) for the EGFP-hSERT plasmid and funding from the Neurological Foundation of New Zealand and Health Research Council of New Zealand.References
- Lyles J, Cadet JL (2003) Methylenedioxymethamphetamine (MDMA,Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Brain Res Rev 42: 155-168.
- Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI (2003) The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharmacol Rev 55: 463-508.
- Howell LL, Kimmel HL (2008) Monoamine transporters and psychostimulant addiction. Biochem Pharmacol 75: 196-217.
- Baumann MH, Wang XY, Rothman RB (2007) 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacology 189: 407-424.
- Sitte HH, Freissmuth M (2010) The reverse operation of Na+/Cl--coupled neurotransmitter transporters - why amphetamines take two to tango. J Neurochem 112: 340-355.
- Qian Y, Galli A, Ramamoorthy S, Risso S , DeFelice LJ, et al. (1997) Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci 17: 45-57.
- Ramamoorthy S, Giovanetti E, Qian Y, Blakely RD (1998) Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J Biol Chem 273: 2458-2466.
- Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S (2005) A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: Evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci 25: 29-41.
- Jayanthi LD, Ramamoorthy S (2005) Regulation of monoamine transporters: Influence of psychostimulants and therapeutic antidepressants. AAPS J 7: E728-E738.
- Ramamoorthy S, Shippenberg TS, Jayanthi LD (2011) Regulation ofmonoamine transporters: Role of transporter phosphorylation. Pharmacol Ther 129: 220-238.
- Kivell B, Day D, Bosch P, Schenk S, Miller J (2010) MDMA causes a redistribution of serotonin transporter from the cell surface to the intracellular compartment by a mechanism independent of phospho-p38-mitogen activated protein kinase activation. Neuroscience 168: 82-95.
- Yasuda K, Itakura M, Aoyagi K, Sugaya T, Nagata E, et al. (2011) PKC Dependent Inhibition of Ca2+-Dependent Exocytosis from Astrocytes. Glia 59: 143-151.
- Nair SG, Gudelsky GA (2004) Protein kinase C inhibition differentially affects 3,4-methylenedioxymethamphetamine-induced dopamine release in the striatum and prefrontal cortex of the rat. Brain Res 1013: 168-173.
- Ramamoorthy S, Samuvel DJ, Buck ER, Rudnick G, Jayanthi LD (2007) Phosphorylation of threonine residue 276 is required for acute regulation of serotonin transporter by cyclic GMP. J Biol Chem 282: 11639-11647.
- Earles C, Schenk JO (1998) Rotating disk electrode voltammetricmeasurements of dopamine transporter activity: An analytical evaluation. Anal Biochem 264: 191-198.
- Thompson AC, Zapata A, Justice JB Jr, Vaughan RA, Sharpe LG, et al. (2000) Kappa-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci 20: 9333- 9340.
- Hagan CE, Neumaier JF, Schenk JO (2010) Rotating disk electrode voltammetric measurements of serotonin transporter kinetics in synaptosomes. J Neurosci Methods 193: 29-38.
- Perez XA, Andrews AM (2005) Chronoamperometry to determine differential reductions in uptake in brain synaptosomes from serotonin transporter knockout mice. Anal Chem 77: 818-826.
- John CE, Jones SR (2007) Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology 52: 1596-1605.
- Hashemi P, Dankoski EC, Petrovic J, Keithley RB, Wightman RM (2009) Voltammetric Detection of 5-Hydroxytryptamine Release in the Rat Brain. Anal Chem 81: 9462-9471.
- Burnette WB, Bailey MD, Kukoyi S, Blakely RD, Trowbridge CG, et al. (1996) Human norepinephrine transporter kinetics using rotating disk electrode voltammetry. Anal Chem 68: 2932-2938.
- Chen N, Trowbridge CG, Justice JB Jr (1999) Cationic modulation of human dopamine transporter: Dopamine uptake and inhibition of uptake. J Pharmacol Exp Ther 290: 940-949.
- Hagan CE, Schenk JO, Neumaier JF (2011) The Contribution of Low-Affinity Transport Mechanisms to Serotonin Clearance in Synaptosomes. Synapse 65: 1015-1023.
- Oz M, Libby T, Kivell B, Jaligam V, Ramamoorthy S, et al. (2010) Real-time, spatially resolved analysis of serotonin transporter activity and regulation using the fluorescent substrate, ASP(+). J Neurochem 114: 1019-1029.
- Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD (2005) p38 MAPK activation elevates serotonin transport activity via a traffickingindependent, protein phosphatase 2A-dependent process. J Biol Chem 280: 15649-15658.
- Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S (2005) Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol 67: 2077-2087.
- Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA, et al. (2000) Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J Neurosci 20: 7571-7578.
- Kramer HK, Poblete JC, Azmitia EC (1997) Activation of protein kinase C (PKC) by 3,4-methylenedioxymethamphetamine (MDMA) occurs through the stimulation of serotonin receptors and transporter. Neuropsychopharmacology 17: 117-129.
- Kramer HK, Poblete JC, Azmitia EC (1998) Characterization of the translocation of protein kinase C (PKC) by 3,4-methylenedioxymethamphetamine (MDMA/ Ecstasy) in synaptosomes: Evidence for a presynaptic localization involvingthe serotonin transporter (SERT). Neuropsychopharmacology 19: 265-277.
- Zhu CB, Hewlett WA, Feoktistov I, Biaggioni I, Blakely RD (2004) Adenosine receptor, protein kinase G and p38 mitogen-activated protein kinasedependent up-regulation of serotonin transporters involves both transporter trafficking and activation. Mol Pharmacol 65: 1462-1474.
- Vaughan RA (2004) Phosphorylation and regulation of psychostimulantsensitive neurotransmitter transporters. J Pharmacol Exp Ther 310: 1-7.
- Amphoux A, Vialou V, Drescher E, Bruss M, La Cour CM, et al.(2006) Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 50: 941-952.
- Daws LC (2009) Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther 121: 89- 99.
- Mortensen OV, Kristensen AS, Rudnick G, Wiborg O (1999) Molecular cloning, expression and characterization of a bovine serotonin transporter. Brain Res Mol Brain Res 71: 120-126.
- Engel K, Zhou MY, Wang J (2004) Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem 279: 50042- 50049.
- Dahlin A, Xia L, Kong W, Hevner R, Wang J (2007) Expression and immunolocalization of the plasma membrane monoamine transporter in the brain. Neuroscience 146: 1193-1211.
- Boudanova E, Navaroli DM, Melikian HE (2008) Amphetamine-induced decreases in dopamine transporter surface expression are protein kinase C-independent. Neuropharmacology 54: 605-612.
- Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME (2005) Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. J Biol Chem 280: 10914-10919.
- Cowell RM, Kantor L, Hewlett GH, Frey KA, Gnegy ME (2000) Dopamine transporter antagonists block phorbol ester-induced dopamine release and dopamine transporter phosphorylation in striatal synaptosomes. Eur J Pharmacol 389: 59-65.