
Journal of Andrology & Gynaecology
Download PDF
Review article
*Address for Correspondence: Dr. Grishina Irina Ph.D., Langone Medical Center, New York University and Department of Biology, City College of Technology, City University of New York; E mail: Grishina.irina@gmail.com
Citation: Grishina I. Planar Polarity Factors as Potential Tumor Suppressors in the Urogenital Tract. J Androl Gynaecol. 2014;2(2): 6.
Copyright © 2014 Grishina I. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Andrology & Gynaecology | ISSN 2332-3442 | Volume: 2, Issue: 2
Submission: 17 April 2014 | Accepted: 27 June 2014 | Published: 30 June 2014
Signaling pathways, which function in defining the shape and interactions between epithelial cells also play a role in maintaining tissue integrity, and can affect the progression of benign hyperplasias and cancers. For instance, disruption of the apical/basal (A/B) epithelial polarity can promote epithelial to mesenchymal transition, which is the essential step in cancer microinvasion. Recent studies uncovered that disruptions in the epithelial polarity, perpendicular to the A/B axis, known as the Planar Cell Polarity (PCP), can lead to serious pathohistological abnormalities, including hyperplastic and neoplastic disorders. In this review, I will summarize the recent advances in our understanding of the PCP effects in organ development and disease, in particular, in relation to the reproductive and urogenital tracts, and the prostate gland.
Planar Polarity Factors as Potential Tumor Suppressors in the Urogenital Tract
Irina Grishina*
- Langone Medical Center, New York University and Department of Biology, City College of Technology, City University of New York
*Address for Correspondence: Dr. Grishina Irina Ph.D., Langone Medical Center, New York University and Department of Biology, City College of Technology, City University of New York; E mail: Grishina.irina@gmail.com
Citation: Grishina I. Planar Polarity Factors as Potential Tumor Suppressors in the Urogenital Tract. J Androl Gynaecol. 2014;2(2): 6.
Copyright © 2014 Grishina I. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Andrology & Gynaecology | ISSN 2332-3442 | Volume: 2, Issue: 2
Submission: 17 April 2014 | Accepted: 27 June 2014 | Published: 30 June 2014
Signaling pathways, which function in defining the shape and interactions between epithelial cells also play a role in maintaining tissue integrity, and can affect the progression of benign hyperplasias and cancers. For instance, disruption of the apical/basal (A/B) epithelial polarity can promote epithelial to mesenchymal transition, which is the essential step in cancer microinvasion. Recent studies uncovered that disruptions in the epithelial polarity, perpendicular to the A/B axis, known as the Planar Cell Polarity (PCP), can lead to serious pathohistological abnormalities, including hyperplastic and neoplastic disorders. In this review, I will summarize the recent advances in our understanding of the PCP effects in organ development and disease, in particular, in relation to the reproductive and urogenital tracts, and the prostate gland.
Planar epithelial polarity is controlled by at least two partially independent pathways (Figure 1). One, is transmitted by a set of so called “core PCP” proteins, consisting of the Frizzled (Fzd) receptors; Vangl1/2 (van Gogh 1 and 2), Celsr1/2/3 transmembrane proteins, and the cytoplasmic proteins Dishevelled (Dvl1, -2 and -3) and Prickle (Pk1 and Pk2). Another is mediated by binding of two transmembrane molecules, Fat (Ft) and Dachous (Ds) on the adjacent cell membranes [1]. Both mechanisms regulate coordinated changes in the cell cytoskeleton, resulting in alterations in cell shape, movement and cilia polarity. When activated by the extracellular ligands, the membrane and cytoplasmic components of the PCP systems form distinct complexes, which become distributed asymmetrically, usually, on the apical side of the epithelial layer. The effects of this intercellular polarization produce macroscopic planar polarity phenomena, for instance, a polarized orientation of the cilia in the epithelial ducts, including the reproductive tracts. The Fat/Ds system is regulated at the intracellular level by the highly conserved tumor-suppressor genes, the atypical cadherins Fat1 and 4, and their partners, Ds1 and 2, which bind to each other at the interface of the neighboring cells [2]. The Ft/Ds complex formation is modulated by the Golgi-associated Four-jointed (Fj) kinase (Figure 1). Fj is expressed in a graded pattern in many polarized tissues, which was linked to the graded distribution of the Fat and Ds components [3]. It was suggested that the Fat/Ds system functions as the long distance PCP component to regulate tissue polarity [3]. The Fat and Ds mouse mutants present abnormalities in the development of multiple organs, including the kidney and gastrointestinal tract [2,4]. Loss of Fat4 results in a disruption of polarized divisions in the developing nephron tubules, due to the defects in organization of the actin cytoskeleton, and leads to the cystic kidney disease [5]. The Fat/Ds system has been linked to progenitor cell regeneration and tumor suppression [6]. Vertebrate Fat cadherins activate the tumorsuppressor Hippo/Lats/Wart kinase pathway (Figure 1), which leads to phosphorylation-inactivation of the YAP and TAZ protooncogenes, by preventing their nuclear entry and induction of the cyclin E and anti-apoptotic proteins. In the absence of a Hippo signal, YAP and TAZ translocate to the nucleus and form transcriptional complexes with a number of factors, including beta-catenin, to promote cell division, survival, and migratory and invasive cell properties.
The Fat/Ds system is regulated at the intracellular level by the highly conserved tumor-suppressor genes, the atypical cadherins Fat1 and 4, and their partners, Ds1 and 2, which bind to each other at the interface of the neighboring cells [2]. The Ft/Ds complex formation is modulated by the Golgi-associated Four-jointed (Fj) kinase (Figure 1). Fj is expressed in a graded pattern in many polarized tissues, which was linked to the graded distribution of the Fat and Ds components [3]. It was suggested that the Fat/Ds system functions as the long distance PCP component to regulate tissue polarity [3]. The Fat and Ds mouse mutants present abnormalities in the development of multiple organs, including the kidney and gastrointestinal tract [2,4]. Loss of Fat4 results in a disruption of polarized divisions in the developing nephron tubules, due to the defects in organization of the actin cytoskeleton, and leads to the cystic kidney disease [5]. The Fat/Ds system has been linked to progenitor cell regeneration and tumor suppression [6]. Vertebrate Fat cadherins activate the tumorsuppressor Hippo/Lats/Wart kinase pathway (Figure 1), which leads to phosphorylation-inactivation of the YAP and TAZ protooncogenes, by preventing their nuclear entry and induction of the cyclin E and anti-apoptotic proteins. In the absence of a Hippo signal, YAP and TAZ translocate to the nucleus and form transcriptional complexes with a number of factors, including beta-catenin, to promote cell division, survival, and migratory and invasive cell properties.
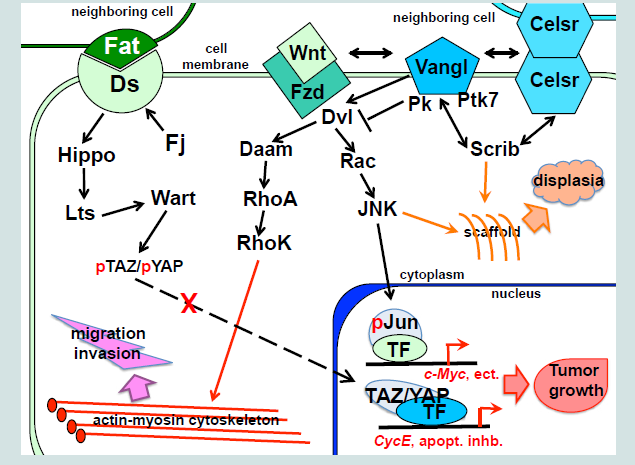
Figure 1: Planar polarity mechanisms in vertebrates: the Ft/Ds and the core PCP pathways.
Loss of FAT function contributes to many types of tumors, including the oral, breast, lung, pancreatic and gastric cancers, melanomas and hepatocellular carcinomas [7]. High levels of YAP1 protein correlate with cancer progression, and poor prognosis for the bladder, prostate, ovarian and uterine cancer patients [8-12]. In turn, irradiation induced apoptosis of prostate cancer cells has been linked to a decrease in the levels of the YAP1 protein and a consequent raise in the BAX cell death factor [12]. Both YAP and TAZ proteins have been implicated in cancer cell migration and metastasis [8,13,14]. The known antagonists of the Hippo pathway include the cilia associated protein NPHP4 [15] and the integrin-associated kinase, which mediate inhibition of Lats kinases upstream of the YAP/TAZ phosphorylation. The Lats 1/2 expression levels are downregulated in many advanced and metastatic cancers, including in prostate cancer, and this effect has been linked to cancer cell resistance to the attachment-dependent apoptosis [16].
The core PCP pathway can be activated by most Wnt ligands, but has a preference for Wnt5a, Wnt9 and Wnt11, which have been designated as “non-canonical” Wnts. Although the Ft/Ds system is considered as the long distance PCP component, the core PCP pathway also has long distance capabilities, enabled by the diffusion of the Wnt ligands [17]. Formation of the Wnt/Fzd complexes is the first step in both the core PCP and Wnt/beta-catenin pathways. Thus, the former is often referred to as the “non-canonical Wnt” signaling. Assembly of the Fzd and Vangl receptor complexes on the apical cell surface appears to favor the PCP pathway [18]. In contrast to the canonical Wnt signaling, the core PCP pathway does not affect nuclear localization of beta-catenin. Furthermore, the core PCP signaling routinely antagonizes the canonical Wnt pathway, first, by recruiting Dvl to the membrane Vangl2 complexes, and also by sequestering other shared components (Wnts, Fzd and Frodo) and promoting the degradation of beta-catenin [19]. The canonical Wnt/beta-catenin signaling is often upregulated in cancers. Therefore, part of the PCP tumor suppressor role is likely exercised simply by suppressing the canonical Wnt targets, while another part is associated with its role in the maintenance of tissue polarity and cohesion.
Activation of the membrane core PCP complexes, is initiated by Wnt binding to Fzd, and few other receptors, such as the Ror [20], and involves active participation of the Vangl1/2 transmembrane complexes, and atypical cadherins, Celsr1/2/3/Flamingo(Figure 1) . Celsr1/2/3 are seven-pass transmembrane proteins [21] with homology to the G protein-coupled receptors. Celsr extracellular region contains an EGF-like and a laminin-G motifs, and nine protocadherin repeats [22]. This combination of domains is expected to promote Celsr signaling following self-recognition across neighboring cells, or upon heterophilic ligand/receptor binding [23]. Vertebrate Vangl1/2 protein products are homologues of the Drosophila Strabismus/Van Gogh gene, and consist of two transmembrane domains and a PDZbinding domain. Vangl2 is essential for the kidney [24] , prostate and genital development (our unpublished data), and patterning of the female genital tract [25].
Membrane PCP signals are transmitted by at least three cytoplasmic factors with links to the cellular cytoskeleton, the Dvl1/2/3, Pk1/2 and Scribble (Scrib), and at least three phosphorylation pathways, regulated by the Rho kinase (RhoK), c-Jun N-terminal kinase (JNK) and the protein tyrosine kinase 7 (Ptk7) (Figure 1).
Dvl1/2/3 are large multidomain proteins that function at the major crossroad between the canonical and non-canonical Wnt pathways (Figure 1)[26]. Dvl possess the N-terminal DIX (Dishevelled, Axin) domain, a PDZ domain, the C-terminal DEP domain (Dvl, Egl-10, Pleckstrin) [27], and two other conserved domains, a basic region and a proline-rich region [28]. Dvl protein can shuttle between the cytoplasm and the nucleus, and its nuclear localization is required for the canonical Wnt signaling [29]. When recruited to the Vangl1/2 complexes, Dvl1/2/3 function as scaffold proteins, linking Vangl1/2 with the downstream signaling components that mediate protein-protein interactions and/or phosphorylation pathways, and induce planar cell polarity and directed movement [30]. Abnormal upregulation of the Dvl 1/2/3 genes in cancers has been linked to enhanced cell invasion [16]. In the cytoplasm, Dvl1/2/3 associate with the Rho GTPase via the formin-homology proteins, Daam1/2 (Dvl-associated activators of morphogenesis). The Dvl/Rho GTPase complexes activate the Rho kinase to induce remodeling of the actin cytoskeleton [31]. All three Dvl genes are expressed in the urogenital ducts and gonads in both sexes, and Dvl deregulation, concomitant with restructuring of the canonical and non-canonical Wnt pathways, has been implicated in the bladder, prostate, testicular, uterine and ovarian cancers.
The core PCP mechanism is involved in differentiation and homeostasis in many epithelial tissues, including the kidney nephrons, and the respective urogenital and reproductive ducts in males and females [25,32-37]. Analysis of mouse fetal lungs and kidneys in the Celsr1-/-, Vangl2-/- or Scrib-/- mutants [24,36,37] uncovered that loss of any of these PCP components results in defects in the cell contacts, linked to the disruptions in the actin-myosin cytoskeleton. Very similar histological defects are observed in the precancerous prostate lesions in human disease, and in the mouse model, heterozygous for the functional Scrib allele [34]. Considering the prevalence of malignant prostate disease in men, it is important to further elucidate the functions of the PCP mechanisms in the maintenance and integrity of the adult prostate, and in the progression of neoplasias.
Scrib is a large scaffold protein, which contains several protein interaction domains, including a leucine-rich region and four PDZ domains [38-40]. In human tissues, SCRIB has been detected in a variety of protein complexes, including with VANGL2, the extracellular signal-regulated kinase (ERK), the tight junction component, ZO-2, and the adenomatous polyposis coli (APC) tumor suppressor complex [34,41]. Scrib has been implicated in diverse cell functions, including cell migration, A/B polarity, and stem cell maintenance. Scrib represents one of the links between the core PCP and Ft/Ds mechanisms. In Drosophila, Scrib acts to regulate activity of the Warts kinase upstream of the Yorkie factor [42], the vertebrate YAP/TAZ homologue. In vertebrate systems, Scrib is a component of the core PCP pathway that genetically interacts with Vangl2 [25,43,44]. Scrib participates in the formation of both the adherens and tight junctions by attracting several adhesion proteins, including beta-catenin, E-cadherin, and the tight junction components, ZO-1 and ZO-2 [37,45,46]. Analysis of the Scrib null murine fetal lungs revealed abnormal shaping of the ducts and disorganized epithelium, associated with significant perturbation of the actin–myosin cytoskeleton and reduced cellular cohesion [37]. These defects were linked to a reduced activity of the RhoA pathway and altered cellular distribution of the Vangl2 and Celsr1 complexes.
Scrib was originally identified as a tumor suppressor in the fruit fly Drosophila [47]. Subsequently, reduced SCRIB protein levels and mislocalization have been linked to many human cancers, including of the colon, intestine, breast, cervix and the prostate [34]. The onset of cervical carcinomas, caused by human papillomavirus, was linked to the ubiquitin-mediated degradation of SCRIB by the viral E6 protein [39,40]. In the mammary epithelium, SCRIB sequestration from cell junctions has been proven sufficient to induce neoplastic transformation, mediated by the disruption of the Rac signaling pathway, associated with the focal adhesion complexes.
SCRIB is an important tumor suppressor in the male sex-accessory gland, the prostate. Microarray analyses of prostate cancer showed a strong association between SCRIB mislocalization and the parameters of prostate cancer progression, including, prostate-specific antigen levels, tumor stage, Gleason grade, and patient survival [34]. About one third of human prostate cancer cases display deregulated SCRIB protein expression [48], but precisely the mislocalization, rather than protein levels, has been associated with poor survival in patients.
Analysis of the Scrib deficient mouse prostates [34] indicates that loss of a single Scrib allele increases the risk of prostate hyperplasia, detected in about 70 % of the mutant males. Hyperplastic cells in the Scrib heterozygous prostates, exhibit keratin 8-positive luminal character and abnormal localization of the E-cadherin, indicating a high probability of a precancerous condition. In turn, loss of both Scrib alleles significantly increases the risk of prostate intraepithelial neoplasias, a precancerous condition. A portion of the Scrib null mice also developed carcinomas of the lungs and pancreas, and dysplastic lesions in the testes and ovaries [34].
Studies to date indicate that in the prostate Scrib functions by negatively regulating a number of mitogen-activated protein kinase (MAPK) cascades. A detailed study in human keratinocytes uncovered that SCRIB downregulation leads to a raise in the Ras kinase activity, which is a major oncogenic pathway in the epithelium of the urethra, bladder and the prostate [49] . Hyperplasia in the murine Scrib heterozygous prostates was linked to increased ERK and MEK kinase activities [34]. Furthermore, at the early stages of prostate cancer, progression could be reversed by pharmacological inhibition of MEK [34], pointing to a direction for the development of clinical treatments.
To date, limited yet intriguing data has been accumulated on the clinical and molecular effects of the PCP components in cancer progression. For instance, microarray analyses show that malignant progression of multiple forms of cancer is associated with decreased levels of VANGL2 (http://www.nextbio.com). Membrane localization of Vangl2 requires an intact microtubule scaffold, while a considerable portion of cytoplasmic Vangl2 co-localize with trafficking vesicles of the Golgi network, aligned along the microtubules [50]. Interestingly, VANGL2 has been shown to regulate vesicle trafficking of many protein components, including metalloproteinases (MMP) [51]. Vangl2 knockdown cells exhibit higher activity of the secreted and membrane bound MMP2 and MMP14, resulting in decreased cellsurface fibronectin, and enhanced migratory and invasive properties.
Recent studies indicate that CELSR1 expression is misregulated by various mechanisms, including alterations in the gene copy number and methylation, in diverse types of human cancers, including lymphomas [52][53], invasive ductal carcinoma of the breast [54], hepatocellular carcinoma [55] and colorectal cancers [56] . Of special interest to the urogenital field, a genome-wide RNAinterference screen identified CELSR1 as an androgen-dependent target, downregulated in metastatic and castration resistant prostate cancer cell line models [57].
The Rho, JNK and Ptk7 are the major phosphorylation pathways downstream of the core PCP signaling. The Rho components are important for epithelial morphogenesis, in particular, for proper ductal branching in the lungs, kidneys and the prostate [36][58]. In the lungs, treatment with a commercial Rho kinase inhibitor Y-27632 (Sigma) phenocopies the branching defects, resulting from the loss of ever Celsr1 or Vangl2 [36]. In turn, treatment of the Celsr1-/- or Vangl2-/- mutant lungs with an activator of the Rho GTPase (upstream of the RhoK), results in increased branching. Our analysis of the murine fetal prostate explant cultures showed that pharmaceutical inhibition of the RhoK results in a smaller number of large prostate buds, thereby inhibiting ductal branching but not the growth process. These results establish a role for the Rho GTPase/kinase pathway in the regulation of epithelial bud formation, a process that includes aspects similar to the epithelial to mesenchymal transition in cancer microinvasion. Importantly, the Rho pathway modulators, tested in these laboratory studies, can be used as a basis in the future clinical trials to impede metastatic progression of cancers.
In Drosophila, JNK kinase, activated downstream of Frizzled and Dvl, is the major mediator in the planar cell polarity signaling. In vertebrates, JNK kinases, encoded by the JNK1/2/3 genes also function in the core PCP pathway. JNK activity requires the Gαo protein, which propagates Wnt/Fzd signal downstream through Dvl1 and Dvl3 [59]. Downstream of Dvl, activation of the small GTPases RhoA, Rac1 and Cdc42, links the mitogen-activated protein kinase kinase (MEKK) cascades to JNK (Figure 1 and [59]). JNK cooperates with Gsk3β to regulate Dvl-mediated stabilization of the microtubule scaffold [60]. In this role, JNK phosphorylates microtubule-associated proteins, MAP2 and MAP1B, resulting in changes in microtubule dynamics. JNK nuclear function is mediated by the transcription factors, c-Jun and ATF2, which cooperate with a number of other signaling pathways to induce cell survival and proliferation [61,62]. Phosphorylated c-Jun negatively regulates cell senescence by suppressing transcription of p53 and cooperates with activated c-Ras to promote oncogenic transformation and metastasis. However, JNK also functions in the stress-induced apoptotic pathways, mediated by Bcl-2. The later underlines the tumor-suppressive JNK function, which can manifest downstream of the oncogenic Ras, or in a context of deficiency for Pten, the universal tumor suppressor in the Akt pathway. Sequencing analyses of cancer genomes revealed clustering of mutations in multiple genes of the JNK pathway in several subtypes of the lung, ovarian, prostate and colorectal cancers [63,64]. In mice models, loss of Jnk1 results in increased risk for skin papilomas, while Jnk1+/-;Jnk2-/- animals develop bronchial lung tumors at high frequency, and Jnk2-/- animals are highly prediposposed to prostate cancer in the Pten+/- background. However, Jnk deficient animals are more resistant to the chemically-induced liver cancer, probably due to a decreased expression of the protooncogenic Jnk targets, c-myc and cyclin E [62]. These observations underline the tissue specificity of the cancer pathways.
Ptk7 kinase has been identified as a component of the vertebrate core PCP pathway, genetically interacting with Vangl2 [65]. Experimental data indicates that Ptk7 can function as a Wnt coreceptor together with Fzd to activate the core PCP pathway, while inhibiting the canonical Wnt/beta-catenin signaling [66,67]. Together with Vangl2, Ptk7 functions to recruit Dvl to the plasma membrane in a process, mediated by the common protein interactor, Rack1(receptor of activated protein kinase C [67]. Shedding of the Ptk7 cytoplasmic domain is a prominent attribute of transformed cells and cancer invasion, and is regulated by the matrix metalloproteinases and the γ-secretase complex. Cleaved Ptk7 localizes to the nucleus and can enhance proliferation, migration and anchorage-independent cancer cell growth [68]. Ptk7 is deregulated in multiple types of human cancer, including in adenocarcinomas of the colon, lungs, breast, kidney and prostate, melanomas and liposarcomas [68,69]. In the lung cancer, Ptk7 acts as a tumor suppressor by inhibiting phosphorylation of the ERK kinase and the AKT [69], both pathways also important in prostate cancerogenesis [34]. Furthermore, recent proteomic analysis identified Ptk7 as a signature for tumor aggressiveness in prostate cancer [70].
In summary, recent clinical data, and studies in model systems strongly indicate that deficiencies in the epithelial planar polarity mechanisms, mediated by the Ft/Ds and core PCP pathways, contribute to tumorigenesis at many stages of cancer progression (Figure 1). Mild defects in the PCP complexes, such in the SCRIB heterozygous and YAP deficient tissues, lead to a loss of epithelial polarity and cohesion, and a bypass in the adhesion-dependent apoptosis, thereby creating a permissive environment for tissue overgrowth at the precancerous and early cancer stages. At carcinoma in situ stage, disruptions in the membrane PCP complexes are transmitted to the JNK and YAP transcriptional targets to induce a number of mitogenic and survival factors. At the advanced cancer stages, and during metastasis, a number of cytoskeleton-connected mediators (TAZ, SCRIB, RhoK, Rac and JNK) are recruited by numerous cancers to promote epithelial to mesenchymal transition, cell motility and podia formation by regulating polymerization of the cytoskeleton. In the last decade, studies in various animal models have begun to unravel the molecular mechanisms that regulate cell polarity during organ development and in cancerogenesis. However, many aspects of the PCP phenomena, for instance, the interactions between the Ft/Ds and the core PCP signaling, and the multilevel regulation mechanisms in each pathway, require further clarification. Thus, it is important that further studies are implemented using the conditional and tissue specific mouse genetic systems to define the effects of the individual and composite PCP mutations at each cancer stage. These studies can potentially provide us with biomedical tools to restrict disease, both at the early and late stages, by modulating the polarity mechanisms to counteract disruptions in the cancer cell cytoskeleton.
References
- Karner C, Wharton KA Jr, Carroll TJ (2004) Planar cell polarity and vertebrate organogenesis. Semin Cell Dev Biol 17: 194-203.
- Saburi S, Hester I, Goodrich L, McNeill H (2012) Functional interactions between Fat family cadherins in tissue morphogenesis and planar polarity. Development 139: 1806-1820.
- Sharma P, McNeill H (2013) Regulation of long-range planar cell polarity by Fat-Dachsous signaling. Development 140: 3869 - 3881.
- Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, et al. (2011) Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 138: 947-957.
- Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, et al. (2008) Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet 40: 1010-1015.
- Sadeqzadeh E, de Bock CE, Thorne RF (2014) Sleeping Giants: Emerging Roles for the Fat Cadherins in Health and Disease. Med Res Rev 34: 190- 221.
- Katoh M (2012) Function and cancer genomics of FAT family genes (review). Int J Oncol 41: 1913-1918.
- Cai H, Xu Y (2013) The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal 11: 31.
- Liu JY, Li YH, Lin HX, Liao YJ, Mai SJ, et al. (2013) Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder. BMC Cancer 13: 349.
- Liu T, Liu Y, Gao H, Meng F, Yang S, et al. (2013) Clinical significance of yes-associated protein overexpression in cervical carcinoma: the differential effects based on histotypes. Int J Gynecol Cancer 23:735-742.
- Xia Y, Chang T, Wang Y, Liu Y, Li W, et al. (2014) YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PLoS One 9: e91770.
- Zagurovskaya M, Shareef MM, Das A, Reeves A, Gupta S, et al. (2009) EGR- 1 forms a complex with YAP-1 and upregulates Bax expression in irradiated prostate carcinoma cells. Oncogene 28: 1121-1131.
- han SW, Lim CJ, Guo K, Ng CP, Lee I, et al. (2008) A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 68: 2592-2598.
- Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, et al. (2014) Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J 33: 468-481.
- Habbig S, Bartram MP, Müller RU, Schwarz R, Andriopoulos N, et al. (2011) NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J Cell Biol 193: 633-642.
- Zhao Y, Yang ZQ, Wang Y, Miao Y, Liu Y, et al. (2010) Dishevelled-1 and dishevelled-3 affect cell invasion mainly through canonical and noncanonical Wnt pathway, respectively, and associate with poor prognosis in non-small cell lung cancer. Mol Carcinog 49: 760-770.
- Gao B, Song H, Bishop K, Elliot G, Garrett L, et al. (2011) Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev Cell 20: 163-176.
- Veeman MT, Axelrod JD, Moon RT (2003) A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell 5: 367- 377.
- Weidinger G, Moon RT (2003) When Wnts antagonize Wnts. J Cell Biol 162: 753-755.
- He F, Xiong W, Yu X, Espinoza-Lewis R, Liu C, et al. (2008) Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development 135: 3871-3879.
- Shima Y, Copeland NG, Gilbert DJ, Jenkins NA, Chisaka O, et al. (2002) Differential expression of the seven-pass transmembrane cadherin genes Celsr1-3 and distribution of the Celsr2 protein during mouse development. Dev Dyn 223: 321- 332.
- Usui T, Shima Y, Shimada Y, Hirano S, Burgess RW, et al. (1999) Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 98: 585-595.
- Strutt H, Strutt D (2008) Differential stability of flamingo protein complexes underlies the establishment of planar polarity. Curr Biol 18: 1555-1564.
- Yates LL, Papakrivopoulou J, Long DA, Goggolidou P, Connolly JO, et al. (2010b)The planar cell polarity gene Vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum Mol Genet 19: 4663-4676.
- Vandenberg AL, Sassoon DA (2009) Non-canonical Wnt signaling regulates cell polarity in female reproductive tract development via van Gogh-like 2. Development 136: 1559-1570.
- Shafer B, Onishi K, Lo C, Colakoglu G, Zou Y (2011) Vangl2 promotes Wnt/ Planar Cell Polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev Cell 20: 177-191.
- Boutros M, Mlodzik M (1999) Dishevelled: at the crossroads of divergent intracellular signaling pathways. Mech Dev 83: 27-37.
- Gao C, Chen YG (2010) Dishevelled: The hub of Wnt signaling. Cell Signal 22: 717 - 727.
- Itoh K, Brott BK, Bae GU, Ratcliffe MJ, Sokol SY (2005) Nuclear localization is required for Dishevelled function in Wnt/beta-catenin signaling. J Biol 4: 3.
- Wallingford JB, Habas R (2005) The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development 132: 4421-4436.
- Habas R, Kato Y, He X (2001) Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107: 843-854.
- Mericskay M, Kitajewski J, Sassoon D (2004) Wnt5a is required for proper epithelial-mesenchymal interactions in the uterus. Development 131: 2061-2072.
- Miller C, Sassoon DA (1998) Wnt-7a maintains appropriate uterine patterning during the development of the mouse female reproductive tract. Development 125: 3201-3211.
- Pearson HB, Perez-Mancera PA, Dow LE, Ryan A, Tennstedt P, et al. (2011) SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice pr006Fmotes prostate neoplasia. J Clin Invest 121: 4257- 4267.
- Yamaguchi TP, Bradley A, McMahon AP, Jones SA (1999) Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126: 1211-1223.
- Yates LL, Schnatwinkel C, Murdoch JN, Bogani D, Formstone CJ, et al. (2010a) The PCP genes Celsr1 and Vangl2 are required for normal lung branching morphogenesis. Hum Mol Genet 19: 2251-2267.
- Yates LL, Schnatwinkel C, Hazelwood L, Chessum L, Paudyal A, et al. (2013) Scribble is required for normal epithelial cell-cell contacts and lumen morphogenesis in the mammalian lung. Dev Biol 373: 267-280.
- Bilder D, Perrimon N (2000) Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 403: 676 - 680.
- Nakagawa S, Huibregtse JM (2000) Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol 20: 8244-8253.
- Nakagawa S1, Yano T, Nakagawa K, Takizawa S, Suzuki Y, et al. (2004) Analysis of the expression and localisation of a LAP protein, human scribble, in the normal and neoplastic epithelium of uterine cervix. Br J Cancer. 90:194- 199.
- Metais JY, Navarro C, Santoni MJ, Audebert S, Borg JP (2005) hScrib interacts with ZO-2 at the cell-cell junctions of epithelial cells. FEBS Lett 579: 3725-3730.
- Verghese S, Waghmare I, Kwon H, Hanes K, Kango-Singh M (2012) Scribble acts in the Drosophila fat-hippo pathway to regulate warts activity. PLoS One 7: e47173.
- Montcouquiol M, Kelley MW (2003) Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. J Neurosci 23: 9469- 9478.
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, et al. (2003) Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423: 173 - 177.
- Navarro C, Nola S, Audebert S, Santoni MJ, Arsanto JP, et al. (2005) Junctional recruitment of mammalian Scribble relies on E-cadherin engagement. Oncogene 24: 4330 - 4339.
- Sun Y, Aiga M, Yoshida E, Humbert PO, Bamji SX (2009) Scribble interacts with beta-catenin to localize synaptic vesicles to synapses. Mol Biol Cell 20: 3390 - 3400.
- Bilder D, Li M, Perrimon N (2000) Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289: 113-116.
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18: 11- 22.
- Nagasaka K1, Pim D, Massimi P, Thomas M, Tomaić V, et al. (2010) The cell polarity regulator hScrib controls ERK activation through a KIM sitedependent interaction. Oncogene 29: 5311-5321.
- Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, et al. (2010) Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol 12: 41-64.
- Cantrell VA, Jessen JR (2010) The planar cell polarity protein Van Gogh- Like 2 regulates tumor cell migration and matrix metalloproteinase-dependent invasion. Cancer Lett 287: 54-61.
- Del Giudice I, Messina M, Chiaretti S, Santangelo S, Tavolaro S, et al. (2012) Behind the scenes of non-nodal MCL: downmodulation of genes involved in actin cytoskeleton organization, cell projection, cell adhesion, tumour invasion, TP53 pathway and mutated status of immunoglobulin heavy chain genes. Br J Haematol 156: 601-611.
- Kaucká M, Plevová K, Pavlová S, Janovská P, Mishra A, et al. (2013) The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res 73: 1491- 1501.
- Liao S, Desouki MM, Gaile DP, Shepherd L, Nowak NJ, et al. (2012) Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosomes Cancer 51: 1067-1078.
- Ammerpohl O, Pratschke J, Schafmayer C, Haake A, Faber W, et al. (2012) Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer 130: 1319-1328.
- Katoh M, Katoh M (2007) Comparative integromics on non-canonical WNT or planar cell polarity signaling molecules: transcriptional mechanism of PTK7 in colorectal cancer and that of SEMA6A in undifferentiated ES cells. Int J Mol Med 20: 405-409.
- Imberg-Kazdan K, Ha S, Greenfield A, Poultney CS, Bonneau R, et al. (2013) A genome-wide RNA interference screen identifies new regulators of androgen receptor function in prostate cancer cells. Genome Res 23: 581- 591.
- Meyer TN, Schwesinger C, Sampogna RV, Vaughn DA, Stuart RO, et al. (2006) Rho kinase acts at separate steps in ureteric bud and metanephric mesenchyme morphogenesis during kidney development. Differentiation 74: 638-647.
- Bikkavilli RK, Feigin ME, Malbon CC (2008) G alpha o mediates WNT-JNK signaling through dishevelled 1 and 3, RhoA family members, and MEKK 1 and 4 in mammalian cells. J Cell Sci 121: 234-245.
- Ciani L, Salinas PC (2007) c-Jun N-terminal kinase (JNK) cooperates with Gsk3beta to regulate Dishevelled-mediated microtubule stability. BMC Cell Biol. 8: 27.
- Blank U, Brown A, Adams DC, Karolak MJ, Oxburgh L (2009) BMP7 promotes proliferation of nephron progenitor cells via a JNK-dependent mechanism. Development 136: 3557-3566.
- Tournier C (2013) The 2 Faces of JNK Signaling in Cancer. Genes Cancer 4: 397-400.
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446: 153- 158.
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, et al. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466: 869-873.
- Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, et al. (2004) PTK7/ CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 430: 93-98.
- Golubkov VS, Chekanov AV, Cieplak P, Aleshin AE, Chernov AV, (2010) The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: implications in cancer and embryogenesis. J Biol Chem 285: 35740-35749.
- Peradziryi H, Tolwinski NS, Borchers A (2012) The many roles of PTK7: a versatile regulator of cell-cell communication. Arch Biochem Biophys 524: 71-76.
- Na HW, Shin WS, Ludwig A, Lee ST (2012) The cytosolic domain of protein-tyrosine kinase 7 (PTK7), generated from sequential cleavage by a disintegrin and metalloprotease 17 (ADAM17) and γ-secretase, enhances cell proliferation and migration in colon cancer cells. J Biol Chem 287: 25001- 25009.
- Kim JH, Kwon J, Lee HW, Kang MC, Yoon HJ, et al. (2014) Protein tyrosine kinase 7 plays a tumor suppressor role by inhibiting ERK and AKT phosphorylation in lung cancer. Oncol Rep 31: 2708-2712.
- Liu Y, Chen J, Sethi A, Li QK, Chen L, et al. (2014) Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol Cell Proteomics Apr 22.