
Advances in Diabetes & Endocrinology
Download PDF
Research Article
*Address for Correspondence: Marc L. Goalstone, Department of Endocrinology, Metabolism and Diabetes, Eastern Colorado Health Care System (Denver VA Medical Center), Mail Stop 151, 1055 Clermont Street, Denver, Colorado 80220, USA, Tel: 303-399-8020; Fax: 303-377-5686; E-mail: Marc.Goalstone@va.gov
Citation: Pott GB, Tsurudome M, Bui J, Goalstone ML. ERK5 and JNK Regulate the Expression of VCAM-1 Differentially in Insulin-and Tumor Necrosis Factor-α Stimulated Rat Aortic Endothelial Cells. Adv Diabetes Endocrinol 2016; 1(1): 6.
Copyright © 2016 Pott GB, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Advances in Diabetes & Endocrinology | Volume: 1, Issue: 1
Submission: 16 September, 2016 | Accepted: 21 October, 2016| Published: 28 October, 2016

In order to accomplish dual transfection experiments we needed to demonstrate that transfection of both short hairpin RNA clones (i.e. shERK5 and shJNK) in the same cells would elicit dual downregulation of each kinase (Figure 2). Here we show that total ERK5 protein was down regulated in the presence of the single transfection of shERK5 and JNK protein was down regulated in the presence of shJNK. Additionally, we demonstrated that the dual transfection of shERK5 and shJNK down regulated both ERK5 and JNK simultaneously.
We have previously demonstrated that Western blot analysis (i.e. total protein content) does not always reflect changes in the amounts of cell surface protein [7]. Thus, we measured changes only in surface VCAM-1 protein by flow cytometry with respect to insulin and/or TNFα stimulation in the absence and presence of shERK5 and shJNK. We first measured VCAM-1 cell surface expression in quiescent cells and cells stimulated with insulin or TNFα alone or in combination in order to determine baseline negative and positive controls, respectively. Insulin alone did not significantly increase surface VCAM-1 expression above that seen for negative controls (Figure 3). In contrast we observed significant (P < 0.05) increases in surface VCAM-1 in the presence of TNFα alone or in combination with insulin as compared to negative controls.
ERK5 and JNK Regulate the Expression of VCAM-1 Differentially in Insulin-and Tumor Necrosis Factor-α Stimulated Rat Aortic Endothelial Cells
Gregory B. Pott1, Mark Tsurudome2, Jamie Bui2 and Marc L. Goalstone1,2*
- 1Eastern Colorado Health Care System, Denver, Colorado, USA
- 2Department of Endocrinology, Metabolism and Diabetes, Eastern Colorado Health Care System (Denver VA Medical Center) and University of Colorado Denver, Denver, Colorado, USA
*Address for Correspondence: Marc L. Goalstone, Department of Endocrinology, Metabolism and Diabetes, Eastern Colorado Health Care System (Denver VA Medical Center), Mail Stop 151, 1055 Clermont Street, Denver, Colorado 80220, USA, Tel: 303-399-8020; Fax: 303-377-5686; E-mail: Marc.Goalstone@va.gov
Citation: Pott GB, Tsurudome M, Bui J, Goalstone ML. ERK5 and JNK Regulate the Expression of VCAM-1 Differentially in Insulin-and Tumor Necrosis Factor-α Stimulated Rat Aortic Endothelial Cells. Adv Diabetes Endocrinol 2016; 1(1): 6.
Copyright © 2016 Pott GB, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Advances in Diabetes & Endocrinology | Volume: 1, Issue: 1
Submission: 16 September, 2016 | Accepted: 21 October, 2016| Published: 28 October, 2016
Abstract
Vascular Cell Adhesion Molecule-1 (VCAM-1) is a cell surface molecule that is expressed on vascular endothelial cells. One of the many functions of VCAM-1 is to interact with serum monocytes, causing them to adhere to the endothelium and transmigrate into the medial portion of the artery. Increased expression of VCAM-1 is part of the pathology of vascular inflammation and atherosclerosis. Insulin and Tumor Necrosis Factor-α (TNFα) are physiological stimulators of cell surface VCAM-1 expression and their intracellular signaling is regulated, in part, by intracellular kinases. We show here that Extracellular Signal-regulate Kinase-5 (ERK5) and c-Jun N-terminal Kinase (JNK) have opposing effects on insulin-and TNFα-stimulated VCAM-1 expression in Rat Aorta Endothelial Cells (RAEC). Using short hairpin ERK5 plasmid DNA and short hairpin JNK plasmid DNA we decreased the expression of ERK5 and JNK, respectively, producing ERK5 Knockdown (KD) and JNK KD cell lines when this hairpin-producing plasmid DNA were expressed in RAEC the following occurred: (1) the simultaneous expression of both ERK KD and JNK KD in the absence of insulin and TNFα increased the basal levels of cell surface VCAM-1, (2) expression of ERK KD decreased insulin and TNFα-stimulated VCAM-1 expression, (3) expression of JNK KD increased the expression of insulin and TNFα-stimulated VCAM-1 and (4) expression of ERK5 KD plus JNK KD stimulated an increase in both insulin and TNFα-stimulated VCAM-1 above that seen for positive controls alone.Keywords
ERK5; JNK; VCAM-1; Insulin; TNFΑ; AtherosclerosisIntroduction
The integrity of the vascular wall is regulated in part by molecular factors in the serum. Hormones, growth factors and cytokines interact with the endothelium and cause physiological and pathophysiological events within the arteries. Atherosclerosis and inflammation are pathologic conditions of the vascular system and manifest in part as neo-intima formation, decreased arterial lumen diameter and occlusion of the arteries. Additionally, changes in the amounts of cell surface and internal proteins via their regulation play important roles in the physiology and the sequelae of pathophysiology of the vasculature.Diabetes is a risk factor for cardiovascular disease. The pathology of diabetes is, but not limited to, increased serum insulin (hyperinsulinemia), glucose (hyperglycemia) and inflammatory cytokines. Hyperinsulinemia and increased presence of Tumor Necrosis Factor-alpha (TNFα) are among the many players of diabetes that contribute to the pathology of atherosclerosis. We and others have demonstrated that increased concentrations of insulin and TNFα stimulate increased total and cell surface expression of Vascular Cell Adhesion Molecule-1 (VCAM-1) [1,2]. The transduction of serum insulin and TNFα information into internal cellular events of the endothelial cells is regulated in part by internal cellular proteins of the kinase super family. Extracellular Signal- Regulated Kinase-5 (ERK5) and c-Jun N-terminal Kinase (JNK) are important kinase mediators of cell surface transduction proteins [3,4]. These two intracellular kinases are also important in cellular activities such as cellular proliferation and inflammation [5].
Here we show that ERK5 and JNK are positive and negative regulators, respectively, of insulin and TNFα stimulation of VCAM-1 expression in Rat Aorta Endothelial Cells (RAEC). Additionally, we report here that there appears to be cross-talk between ERK5 and JNK in the regulation of insulin and TNFα-stimulated surface expression of VCAM-1 on rat aorta endothelial cells.
Methods
MaterialsAll general lab reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary rabbit antibodies to JNK (9258s), ERK5 (3372s), and alpha-tubulin (2144S) were from Cell Signaling (Boston, MA, USA). The primary rabbit antibody to VCAM-1 (NBP1-95622) was from Novus Biologicals (Littleton, CO, USA) and goat anti-rabbit-secondary antibody IRDye680RD (926-68171) was from LI-COR (Lincoln, NE, USA). DyLight 488-conjugated anti- VCAM-1 antibody was from Thermo Fisher Scientific (Pittsburgh, PA, USA). Four-well chamber slides were from Thermo Fisher and DAPI Mounting Medium was from Vector Labs (Burlingame, CA, USA). Rat aorta vascular endothelial cells (RAEC) (CRL-1444) were from ATCC (Manassas, VA, USA) and culture medium was from Life Technologies (Grand Island, NY, USA). JNK short-hairpin RNA (shRNA) (KR43333P) and ERK5 shRNA (K447075P) were obtained from SA Biosciences/Qiagen ThermoFisher (Valencia, CA, USA). Transfection medium (sc108062) and transfection reagent (sc108061) were from Santa Cruz (Dallas, TX, USA).
Cell culturing
RAEC were cultured in Complete Growth Medium (CGM) [DMEM with 4 mM L-glutamine, 4.5 g/L-glucose and 1.5 g/L sodium bicarbonate) and supplemented with 10% Heat-Inactivated Fetal Bovine Serum (HI-FBS) (10438-026) (Life Technologies, Grand Island, NY, USA) and 1% Antimycotic-Antibiotic solution (15240-062) (Life Technologies) and cultured at 37 °C, 5% CO2 atmosphere.
SDS-PAGE and Western blot analysis
Sodium Dodecyl Sulfate Polyacrylamide Electrophoresis (SDS PAGE) was performed using cleared lysates. Western blot analyses were subsequently performed as previously described, with the following differences [6]. After completion of protein transfer, membranes were washed in ultra-pure water for 5 min. Membranes were then incubated in 3% non-fat milk (milk) in Tris-buffered Saline (TBS) blocking solution for 1 hr at room temperature and then incubated with a designated primary antibody solution (1:1000 in 3% milk/TBS-T[0.1% tween]) overnight at 4 °C. Membranes were washed 4 times with TBS plus Tween (TBS-T) for 5 min at room temperature and then incubated with a goat anti-rabbit secondary antibody (1:5000 in 3% milk/TBS-T) conjugated to fluorochrome IR680RD for 1 hourat room temperature. Membranes were washed 4 times with TBS-T for 5 min each time at room temperature and then incubated with a rabbit anti-tubulin primary antibody solution (1:1000 in 3% milk/TBS-T) for 3 hr at room temperature. After washing the membranesfor 4 times with TBS-T, the membranes were allowed to dry before performing densitometry. Densitometry was performed using an Odyssey Licor system (Lincoln, NE, USA). Alpha-tubulin protein was used to normalize VCAM-1 signals.
Preparation of shRNA stable cell lines
RAEC were grown to 50-70% confluence in CGM in 6-well culture plates. Cells were transfected with shERK5 or shJNK inhibitory plasmids as previously described [7]. Cells were incubated in CGM containing 2 µg/mL of Puromycin (Sigma-Aldrich) for 2-3 weeks for selection of Puromycin resistant transformants.
Dual transfection of stable cell lines
To examine the effect of simultaneous ERK5 and JNK knockdown on VCAM-1 expression, the ERK5 shRNA stable cell line (ERK5 KD) was transiently transfected with shJNK plasmids and the JNK shRNA stable cell line (JNK KD) was transiently transfected with the shERK5 plasmid. These two protocols were carried out in order to see if any differences occurred with respect to transfection sequence. Stable cell lines were transiently transfected with shRNA plasmid DNA as described above and incubated for 5 hr with the DNA transfection mix. Subsequently the transfection mix was aspirated and replaced with 2.0 mL CGM with 2 µg/mL puromycin. Stimulation of cells by insulin and/or TNFα occurred 48 hours after transient transfection was accomplished.
Stimulation of VCAM-1 expression
RAEC were cultured in CGM, whereas shRNA stable cell lines (e.g., JNK KD and ERK5 KD) were cultured in CGM containing 2 µg/mL. Puromycin until assays were performed. After incubating the transfected cells for an additional 48 hr the cells were stimulated without or with insulin (10 nM) alone for 1hour or TNFµ (10 ng/mL) alone for six hours or in combination. Thereafter we evaluated for VCAM-1 expression by Western blot analysis and flow cytometry.
Flow cytometry
Mock transfected RAEC (control), stably transfected ERK5 KD RAEC or JNK KD cell lines were inoculated into 6-well tissue culture dishes, transiently transfected with vehicle, shJNK or shERK5 plasmids, respectively, and stimulated with insulin (10 nM) or TNFα (10 ng/mL) alone or combination as described above. The cells were washed twice with 2 mL of 1X PBS (Gibco). The PBS was aspirated and 0.5 mL of Cell Dissociation Solution Non-Enzymatic (Sigma-Aldrich, St. Louis, MO, USA) was added to each well. After incubating the cells at 37 °C and 5% CO2 for 30 min, 1 mL of 1% Bovine Serum Albumin (BSA, Sigma-Aldrich) in PBS was added to the cells and then were gently triturated into a single cell suspension. The cells were transferred to 5 mL Falcon polystyrene round bottom tubes (Thermo Scientific) and centrifuged at 500 x g for 5 min. After aspirating the supernatants, the cells were resuspended in 3 mL 1% BSA, pelleted at 500 x g by centrifugation, and the supernatants were removed by aspiration. The cells were resuspended in 200 µL of 1% BSA. Two microliters of DyLight 488-conjugated anti-VCAM-1 antibody (Life Technologies, Grand Island New York, USA) were added to each tube and the cells were resuspended by vortexing. The cells were incubated in the dark for 30 min at room temperature. The cells were centrifuged, washed twice with 3 mL 1% BSA and resuspended in 200 µL of 1% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA). After incubating the cells for 5 min at room temperature, the cells were diluted with an additional 300 µL of PBS and analyzed using flow cytometry. The experiments were run on a BD LSRII (BD Biosciences, San Jose, CA). MFI and gating percentages as part of data analysis was done using BD FACSDiva v6 software.
Chamber slide cell preparation
2 x 105 of RAEC control (mock transfected), JNK KD or ERK KD stable cell lines were plated separately in 1 mL of CGM in each well of a 4-well chamber slide and allowed to grow for 24 h, 37 °C and 5% CO2. The medium was aspirated and 1.0 mL of fresh CGM was applied to the cells. ERK5 KD and JNK KD cells were mock transfected, or transiently transfected with shJNK or shERK5, respectively, for 48 hours. Cells were then treated with either vehicle, insulin (10 nM) or TNFα (10 ng/mL) alone or in combination. The medium was aspirated and the cells were washed three times with PBS and then incubated in 400 µL of 4% paraformaldehyde in PBS for 30 minutes. The medium was aspirated and washed three times with 1 mL of PBS. The final PBS wash was aspirated and 400 µL of a 1:1000 DyLight anti-VCAM-1 antibody soluton in 1% BSA was added to each chamber and incubated for 30 minutes at room temperature. The cells were then washed three times with 500 µL of 1% BSA. The chamber walls were removed and one drop of DAPI Mounting Medium was added to each group of cells on the slide. Cells were then sealed with a glass cover slip using clear nail polish. Slides were kept in a dark refrigerator until microscopic visualization.
Confocal microscopy
A single, non-confluent monolayer of cells was imaged with a Leica TSC SP8X white light laser scanning confocal microscope (Leica Microsystems GmbH. Ernst-Leitz-Straße 17-37 Wetzlar, 35578 Germany). All image acquisitions were carried out using the Leica Application Suite X (version 1.1.0.12420, LASX AF). Excitation of the DAPI channel was accomplished using a 405 nm diode laser with an excitation intensity level of 2.67%. Emission signal was captured with standard PMT Channel 1 and an emission gap of 430 nm-480 nm. The Leica Supercontinuum white light excitation laser line (488 nm) with 3 % intensity level was used to for Alexa fluor 488. Emission signals were captured with the Leica HyD 2 detector (Hybird 2 PMT) with an emission gap of 505 nm-555 nm.
Data analysis
Data were analyzed by either unpaired Student´s t test (two groups) or ANOVA with subsequent Tukey posttest (several groups) as indicated. A “P” value of less than 0.05 was considered significant. Results were expressed as the mean ± Standard Error of the Mean (SEM) of three or more independent experiments.
Results
We have previously shown that clone #1 (from clones 1-4) of short-hairpin (sh) ERK5 had the best efficacy of downregulating total ERK5 protein [8]. In this report we demonstrate that clone #2 of shorthairpin (sh) JNK was the most potent clone to affect a significant decrease of total JNK protein (Figure 1). Although we have previously shown kinase knock-down studies of ERK5 and p38 in Western blot analyses and flow cytometry, we have not demonstrated the effects of shERK5 and shJNK on insulin-and TNFα-stimulated VCAM-1 cell surface expression in Rat Aorta Endothelial Cells (RAEC) [8].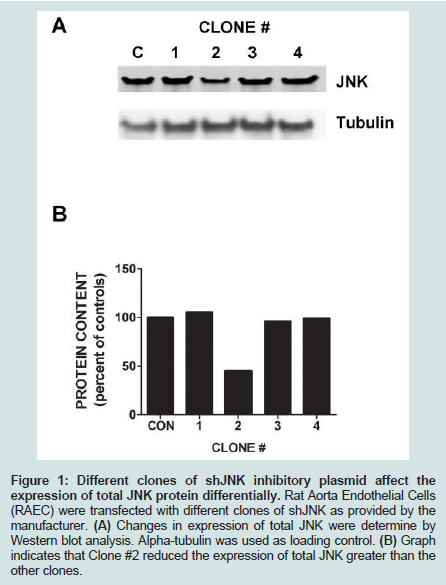
Figure 1: Different clones of shJNK inhibitory plasmid affect the expression of total JNK protein differentially. Rat Aorta Endothelial Cells (RAEC) were transfected with different clones of shJNK as provided by the manufacturer. (A) Changes in expression of total JNK were determine by Western blot analysis. Alpha-tubulin was used as loading control. (B) Graph indicates that Clone #2 reduced the expression of total JNK greater than the other clones.
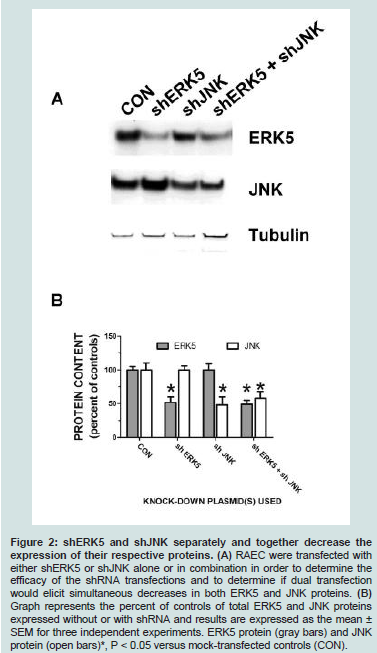
Figure 2: shERK5 and shJNK separately and together decrease the expression of their respective proteins. (A) RAEC were transfected with either shERK5 or shJNK alone or in combination in order to determine theefficacy of the shRNA transfections and to determine if dual transfection would elicit simultaneous decreases in both ERK5 and JNK proteins. (B) Graph represents the percent of controls of total ERK5 and JNK proteins expressed without or with shRNA and results are expressed as the mean ± SEM for three independent experiments. ERK5 protein (gray bars) and JNK protein (open bars)*, P < 0.05 versus mock-transfected controls (CON).
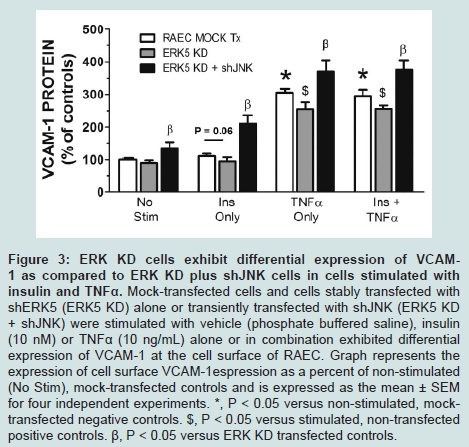
Figure 3: ERK KD cells exhibit differential expression of VCAM-1 as compared to ERK KD plus shJNK cells in cells stimulated with insulin and TNFα. Mock-transfected cells and cells stably transfected withshERK5 (ERK5 KD) alone or transiently transfected with shJNK (ERK5 KD + shJNK) were stimulated with vehicle (phosphate buffered saline), insulin (10 nM) or TNFα (10 ng/mL) alone or in combination exhibited differentialexpression of VCAM-1 at the cell surface of RAEC. Graph represents the expression of cell surface VCAM-1espression as a percent of non-stimulated (No Stim), mock-transfected controls and is expressed as the mean ± SEM for four independent experiments. *, P < 0.05 versus non-stimulated, mocktransfected negative controls. $, P < 0.05 versus stimulated, non-transfected positive controls. β, P < 0.05 versus ERK KD transfected controls.
We then measured changes in surface expression of VCAM-1 in RAEC transfected with shERK5 alone or in cells transfected with shERK5 first and then in combination with shJNK as well as in the absence or presence of insulin or TNFα alone or in their combination (Figure 3). Cells stably transfected with shERK5 (ERK5 KD) and treated with insulin alone exhibited a small, but insignificant (P = 0.06) decrease in cell surface VCAM-1 as compared to positive non-transfected controls. Interestingly, ERK5 KD cells exhibited a significant (P < 0.05) decreased VCAM-1 expression in TNFα and TNFα plus insulin-stimulated cells compared to mock transfected positive controls. In comparison, ERK KD cells transfected with shJNK and then treated with insulin or TNFα alone or in combination exhibited a significant (P < 0.03) increase in cell surface VCAM-1 expression above stimulated positive controls. Interestingly, VCAM-1 expression in non-stimulated ERK5 KD plus shJNK transfected cells was significantly (P < 0.05) greater than that seen in non-stimulated negative controls. These last results suggested that VCAM-1 expression is constitutively down regulated in part by ERK5 and JNK, such that the absence of both kinases and in the absence of stimulation, VCAM-1 expression would be upregulated.
In order to verify that transfection sequence was not an issue, we repeated the above experiments with RAEC transfected with shJNK alone and then followed by transfections with shERK5. In comparison to experiments in Figure 3, we noted a number of interesting differences and analogies in VCAM-1 expression profiles. First, cells transfected with shJNK alone (i.e., JNK KD) and not stimulated with insulin or TNFα, exhibited a slight, but insignificant (P = 0.08) increase in surface VCAM-1 proteins. Second, cells transfected with JNK KD and shERK5 in the absence of stimulus exhibited increased VCAM-1 expression above mock transfected controls suggesting, again, a release of a constitutive negative regulation on VCAM-1 expression. Third, JNK KD cells stimulated with either TNFα alone or in combination with insulin exhibited a significant (P < 0.05) increase in VCAM-1 expression as compared to positive nontransfected controls. Fourth, JNK KD plus shERK5 cells stimulated with insulin or TNFα alone or in combination exhibited a significant (P < 0.05) increase in surface VCAM-1 expression as compared to cells transfected with shJNK alone (Figure 4).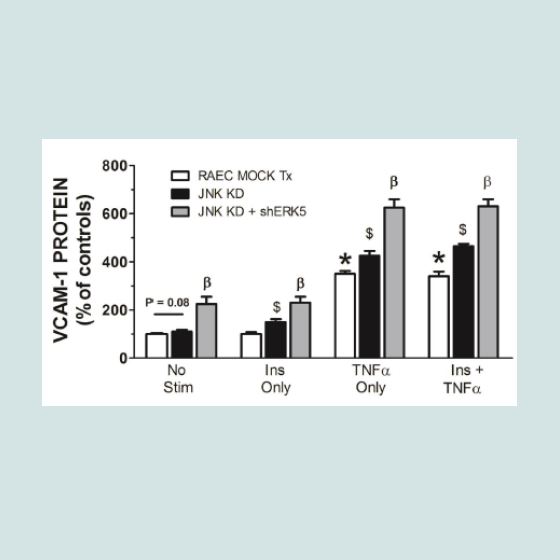
Finally, in order to visualize changes in cell surface expression of VCAM-1 in the presence or absence of insulin and/or TNFα in cells without or with double transfections, we performed immunofluorescence confocal microscopy (Figure 5). Here we observed that the combination of shJNK and shERK5 in RAEC exhibited the greatest increase in VCAM-1 surface expression in the presence of insulin and TNFα.
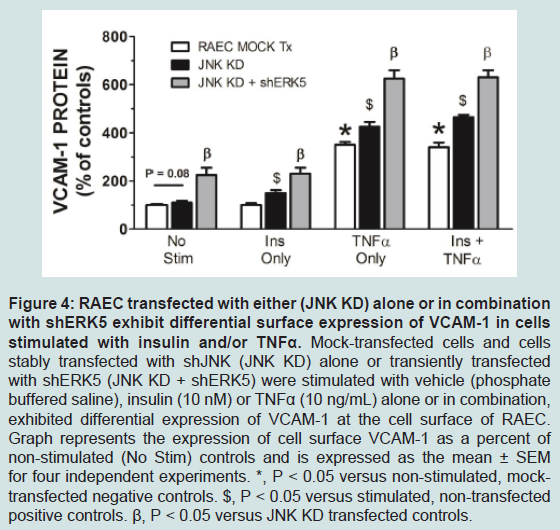
Figure 4: RAEC transfected with either (JNK KD) alone or in combination with shERK5 exhibit differential surface expression of VCAM-1 in cells stimulated with insulin and/or TNFα. Mock-transfected cells and cells stably transfected with shJNK (JNK KD) alone or transiently transfected with shERK5 (JNK KD + shERK5) were stimulated with vehicle (phosphate buffered saline), insulin (10 nM) or TNFα (10 ng/mL) alone or in combination,exhibited differential expression of VCAM-1 at the cell surface of RAEC. Graph represents the expression of cell surface VCAM-1 as a percent of non-stimulated (No Stim) controls and is expressed as the mean ± SEM for four independent experiments. *, P < 0.05 versus non-stimulated, mocktransfected negative controls. $, P < 0.05 versus stimulated, non-transfected positive controls. β, P < 0.05 versus JNK KD transfected controls.
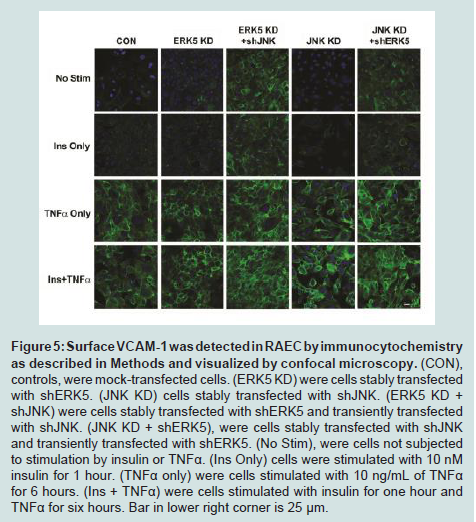
Figure 5: Surface VCAM-1 was detected in RAEC by immunocytochemistry as described in Methods and visualized by confocal microscopy. (CON), controls, were mock-transfected cells. (ERK5 KD) were cells stably transfected with shERK5. (JNK KD) cells stably transfected with shJNK. (ERK5 KD + shJNK) were cells stably transfected with shERK5 and transiently transfected with shJNK. (JNK KD + shERK5), were cells stably transfected with shJNK and transiently transfected with shERK5. (No Stim), were cells not subjected to stimulation by insulin or TNFα. (Ins Only) cells were stimulated with 10 nM insulin for 1 hour. (TNFα only) were cells stimulated with 10 ng/mL of TNFα for 6 hours. (Ins + TNFα) were cells stimulated with insulin for one hour andTNFα for six hours. Bar in lower right corner is 25 μm.
Discussion
We report here that ERK5 and JNK regulate insulin and TNFα-stimulated rat aorta endothelial cell (RAEC) surface VCAM-1 expression. In particular ERK5 appears to be a positive regulator of insulin and TNFα-stimulated VCAM-1 expression whereas JNK seems to be a negative regulator of insulin and TNFα-stimulated VCAM-1 expression.To reiterate, RAEC with less JNK protein (JNK KD and JNK KD + shERK5) exhibited increased VCAM-1 expression even in non-stimulated cells. This suggests that JNK may be an internal negative control of VCAM-1 expression even in quiescent conditions. Second, ERK5 KD transfected cells in the presence of shJNK cells exhibited an increase in VCAM-1 surface expression. This suggests that (1) ERK5 has less influence on VCAM-1 expression than JNK and (2) in the absence of JNK, shERK5 cells express more VCAM-1 because of the activation of other important kinases may be upregulated (Figure 6). Thus, disinhibition of ERK5, along with the disinhibition of JNK appears to cause an even greater expression of VCAM-1.
There have been a vast number of clinical research studies that have measured differences in the amounts of expressed VCAM-1 between control participants versus patients with atherosclerosis. Two such studies have demonstrated that there is no correlation between VCAM-1 expression and vascular disease. Kilic et al. were unable to demonstrate a correlation between aortic stiffness and VCAM- 1 levels [9]. Additionally, Hwang et al. observed that circulation levels of VCAM-1 were not significantly different in Coronary Heart Disease (CHD) patients than that measured in control patients [10].
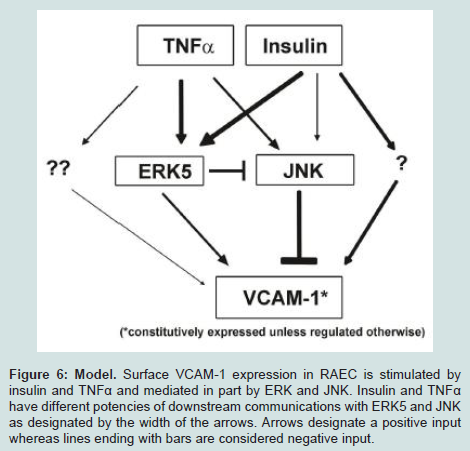
Figure 6: Model. Surface VCAM-1 expression in RAEC is stimulated by insulin and TNFα and mediated in part by ERK and JNK. Insulin and TNFα have different potencies of downstream communications with ERK5 and JNK as designated by the width of the arrows. Arrows designate a positive input whereas lines ending with bars are considered negative input.
In contrast, a number of clinical studies have determined a correlation between vascular inflammation and VCAM-1 expression. Three studies in particular demonstrated a strong association between VCAM-1 expression and atherosclerosis. Mu et al. demonstrated that patients with atherosclerosis expressed an increase in VCAM-1 on endothelial cells and in the tunica media region noted in immunohistochemically stained explanted vascular tissue [11]. Additional studies have shown that there is a strong association between VCAM-1 expression and intimal leukocyte accumulation and atherosclerosis [12,13]. In addition to the latter group, others have demonstrated that introduction of Maritime Pine Bark Extract, Metformin and Atorvastatin have affected a decrease in VCAM-1 expression [14-16]. All of which decreased the detrimental effects of CVD.
There are many clinical research trials that are not favorable to the correlation between atherosclerosis and VCAM-1 expression. Yet, there appears to be a larger number of studies that illustrate a very strong relationship between vascular pathology and VCAM-1 expression. In addition, the uses of both homeopathic and Western medicine applications have indicated that VCAM-1 expression can be regulated. Obviously, more research is needed in this area of human health and pathology.
Cross-talk between internal kinases appears to be a common thread in cellular physiology [17,18]. In some examples, the kinases are redundant in their regulation; that is, both are positive or both are negative regulators. Each is present to ensure that the transduction of outside stimulators is transmitted into the same internal events. In contrast, many kinases have opposing controls of cellular activities, thus modulating many signals to regulate internal events. The transduction of external signals followed by internal kinase modulation is important to the overall “health” of the cell. Balancing the myriad of external signals into coordinated cellular functions is a complicated feat for a cell.
Diabetes is associated with inflammation and atherosclerosis [19,20]. Among the many sequelae of diabetes is insulin resistance and vascular pathophysiology [21,22]. Insulin´s role in inflammation and atherosclerosis is a hotly debated topic [23,24]. In contrast, increased serum and cell surface presence of TNFα is a well-accepted theory of vascular inflammation and increased VCAM-1 expression [4,25].
Our recent studies give thought and evidence that postulate that ERK5 and especially JNK are excellent candidates for molecular therapeutic targets and may be gateways to preventative interventions of vascular inflammation and atherosclerosis.
Acknowledgements
We would like to acknowledge the assistance of Ron Bouchard (Manager of the Microscopy Core) and Harsh Pratap (Flow Cytometry technician for the Mucosal and Vaccine Research Colorado), both of the Eastern Colorado Health Care Service (Denver VAMC). This work was supported by the Research Service of the Department of Veterans Affairs (to MLG) in which Dr. Goalstone is a recipient of a VA Merit Award.References
- Mackesy DZ, Goalstone ML (2011) Insulin augments tumor necrosis factor-alpha stimulated expression of vascular cell adhesion molecule-1 in vascular endothelial cells. J Inflamm 8: 34.
- Lu H, Raptis M, Black E, Stan M, Amar S, et al. (2004) Influence of diabetes on the exacerbation of an inflammatory response in cardiovascular tissue. Endocrinology 145: 4934-4939.
- Zhao M, Liu Y, Bao M, Kato Y, Han J, et al. (2002) Vascular smooth muscle cell proliferation requires both p38 and BMK1 MAP kinases. Arch Biochem Biophys 400: 199-207.
- Inoue K, Zama T, Kamimoto T, Aoki R, Ikeda Y, et al. (2004) TNFalpha-induced ATF3 expression is bidirectionally regulated by the JNK and ERK pathways in vascular endothelial cells. Genes Cells 9: 59-70.
- Jiang Y, Gong XW (2000) Regulation of inflammatory responses by MAPK signal transduction pathways. Sheng Li Xue Bao 52: 267-271.
- Mackesy DZ, Goalstone ML (2014) Extracellular signal-regulated kinase-5: Novel mediator of insulin and tumor necrosis factor α-stimulated vascular cell adhesion molecule-1 expression in vascular cells. J Diabetes 6: 595-602.
- Pott GB, Tsurudome M, Bamfo N, Goalstone ML (2016) ERK2 and Akt are negative regulators of insulin and Tumor Necrosis Factor-α stimulated VCAM-1 expression in rat aorta endothelial cells. J Inflamm 13: 6.
- Pott GB, Tsurudome M, Bui JD, Goalstone ML (2016) ERK5 and p38 Kinase are Positive Regulators of Insulin and TNFα-stimulated VCAM-1 Expression in Rat Aorta Endothelial Cells. Adv Diabetes Endocrinol 1: 7-13.
- Kilic ID, Findikoglu G, Alihanoglu YI, Yildiz BS, Uslu S, et al. (2015) Circulating adhesion molecules and arterial stiffness. Cardiovasc J Afr 26: 21-24.
- Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, et al. (1997) Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation 96: 4219-4225.
- Mu W, Chen M, Gong Z, Zheng F, Xing Q (2015) Expression of vascular cell adhesion molecule-1 in the aortic tissues of atherosclerotic patients and the associated clinical implications. Exp Ther Med 10: 423-428.
- O'Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, et al. (1993) Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest 92: 945-951.
- Davies MJ, Gordon JL, Gearing AJ, Pigott R, Woolf N, et al. (1993) The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol 171: 223-229.
- McGrath KC, Li XH, McRobb LS, Heather AK (2015) Inhibitory effect of a French maritime pine bark extract-based nutritional supplement on TNF-α-induced inflammation and oxidative stress in human coronary artery endothelial cells. Evid Based Complement Alternat Med 2015: 1-7.
- Victor VM, Rovira-Llopis S, Banuls C, Diaz-Morales N, Lopez-Domenech S, et al. (2015) Metformin modulates human leukocyte/endothelial cell interactions and proinflammatory cytokines in polycystic ovary syndrome patients. Atherosclerosis 242: 167-173.
- Tuttolomondo A, Di Raimondo D, Pecoraro R, Maida C, Arnao V, et al. (2016) Early high-dosage atorvastatin treatment improved serum immune-inflammatory markers and functional outcome in acute ischemic strokes classified as large artery atherosclerotic stroke: a randomized trial. Medicine (Baltimore) 95: e3186.
- Campbell M, Allen WE, Silversides JA, Trimble ER (2003) Glucose-induced phosphatidylinositol 3-kinase and mitogen-activated protein kinase-dependent upregulation of the platelet-derived growth factor-β receptor potentiates vascular smooth muscle cell chemotaxis. Diabetes 52: 519-526.
- Nishimoto S, Nishida E (2006) MAPK signalling: ERK5 versus ERK1/2. EMBO Rep 7: 782-786.
- Lebovitz HE (1999) Type 2 diabetes: an overview. Clin Chem 45: 1339-1345.
- Sonne MP, Hojbjerre L, Alibegovic AC, Nielsen LB, Stallknecht B, et al. (2011) Endothelial function after 10 days of bed rest in individuals at risk for type 2 diabetes and cardiovascular disease. Exp Physiol 96: 1000-1009.
- Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL (2009) Inflammation and NF-kappaB in Alzheimer's disease and diabetes. J Alzheimers Dis 16: 809-821.
- Ginsberg HN (2000) Insulin resistance and cardiovascular disease. J Clin Invest 106: 453-458.
- Reddy KJ, Singh M, Bangit JR, Batsell RR (2010) The role of insulin resistance in the pathogenesis of atherosclerotic cardiovascular disease: an updated review. J Cardiovasc Med (Hagerstown) 11: 633-647.
- Aljada A, Saadeh R, Assian E, Ghanim H, Dandona P (2000) Insulin inhibits the expression of intercellular adhesion molecule-1 by human aortic endothelial cells through stimulation of nitric oxide. J Clin Endocrinol Metab 85: 2572-2575.
- Narizhneva NV, Razorenova OV, Podrez EA, Chen J, Chandrasekharan UM, et al. (2005) Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. FASEB J 19: 1158-1160.