
Advances in Diabetes & Endocrinology
Download PDF
Research Article
*Address for Correspondence: Marc L. Goalstone, Department of Endocrinology, Metabolism and Diabetes, Eastern Colorado Health Care System (Denver VA Medical Center), Mail Stop 151, 1055 Clermont Street, Denver, Colorado 80220, USA, Tel: 303.399.8020; Fax: 303.377.5686; E-mail: Marc.Goalstone@ucdenver.edu
Citation: Pott GB, Tsurudome M, Bui J, Goalstone ML. Erk5 and P38 Kinase are Positive Regulators of Insulin and Tnfα - Stimulated Vcam-1 Expression in Rat Aorta Endothelial Cells. Adv Diabetes Endocrinol 2016;1(1): 7.
Copyright © 2016 Pott GB, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Advances in Diabetes & Endocrinology | Volume: 1, Issue: 1
Submission: 23 December, 2015 | Accepted: 15 February, 2016| Published: 20 February, 2016



Since changes in total cellular protein are not always reflected in changes in protein levels at the cell surface, we decided to measure VCAM-1 protein at the cell surface via flow cytometry (Figure 3). Repeating our first experiment in mock transfected RAEC along with stable ERK KD and transiently transfected shp38 cells, we observed that surface VCAM-1 of ERK5 KD cells decreased 28% in the presence of TNFα alone and 39% in the presence of insulin plus TNFα (Figure 3A) as compared to positive controls. In comparison, ERK5 KD plus shp38 cells demonstrate a 32% decrease in VCAM-1 in TNFα stimulated cells and 38% in insulin plus TNFα treated cells.
In a side-by-side comparison to ERK5 KD ± shp38 cells, we determined surface VCAM-1 in cells of p38 KD and p38 KD plus shERK5 (Figure 3B). Interestingly, in insulin-treated p38 KD cells, there was a decrease of 37% VCAM-1 as compared to positive controls, but no statistically significant difference for p38 KD + shERK5 cells. In contrast, cells stimulated with TNFα alone exhibited a decrease in VCAM-1 surface expression of 17% and 36% in p38 KD and p38 KD + shRK5 cells, respectively, while cells stimulated with insulin plus TNFα exhibited a 21% and 23% decrease in surface VCAM-1in p38 KD and p38 KD + shERK5, respectively.

The path from cytokine to intracellular kinase pathway has always been intriguing and this study is no exception. In this study, it appears that the downregulation of both ERK5 and p38 contribute to observed decreases in total and surface VCAM-1 expression in rat aorta endothelial cells. “Cross talk” between different intracellular kinases has profound effects on internal events within the cell. While it appears that the cell has redundant pathways, all are necessary in order to maintain the equilibrium of the cell.
Erk5 and P38 Kinase are Positive Regulators of Insulin and Tnfα - Stimulated Vcam-1 Expression in Rat Aorta Endothelial Cells
Gregory B. Pott1, Mark Tsurudome1, Jamie Bui1 and Marc L. Goalstone1,2*
- 1Eastern Colorado Health Care System, Denver, Colorado, USA
- 2Department of Endocrinology, Metabolism and Diabetes, EasternColorado Health Care System (Denver VA Medical Center) andUniversity of Colorado Denver, Denver, Colorado, USA
*Address for Correspondence: Marc L. Goalstone, Department of Endocrinology, Metabolism and Diabetes, Eastern Colorado Health Care System (Denver VA Medical Center), Mail Stop 151, 1055 Clermont Street, Denver, Colorado 80220, USA, Tel: 303.399.8020; Fax: 303.377.5686; E-mail: Marc.Goalstone@ucdenver.edu
Citation: Pott GB, Tsurudome M, Bui J, Goalstone ML. Erk5 and P38 Kinase are Positive Regulators of Insulin and Tnfα - Stimulated Vcam-1 Expression in Rat Aorta Endothelial Cells. Adv Diabetes Endocrinol 2016;1(1): 7.
Copyright © 2016 Pott GB, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Advances in Diabetes & Endocrinology | Volume: 1, Issue: 1
Submission: 23 December, 2015 | Accepted: 15 February, 2016| Published: 20 February, 2016
Abstract
Type-2 Diabetes Mellitus (T2DM) is a common disease in the Western world. Hyperinsulinemia, hyperlipidemia and vascular inflammation are among the multitude of dysregulations in the Metabolic X Syndrome. Vascular remodeling, occlusion and rupture are effects of the inflammatory events. These events follow cellular increases in expression of cellular adhesion molecules, adhesion of soluble vascular monocytes, their transmigration to the intima of vessels and eventual expression of inflammatory cytokines. Here we show that hyperphysiological concentrations of insulin and tumor necrosis factor-alpha (TNFα) increase both total and cell surface Vascular Cell Adhesion Molecule-1 (VCAM-1) in rat aorta endothelial cells (RAEC) and these actions are in part regulated by the mitogenactivated protein kinases ERK5 and p38. RAEC transfected with RNAi plasmids of ERK5 and p38 and subsequently stimulated with insulin or TNFα alone or in combination exhibited significant decreases in total and cell surface VCAM-1. Western-blot analysis, flow cytometry and confocal microscopy all indicate that ERK5 and p38 MAP kinases are positive regulators of insulin and TNFα stimulated VCAM-1 expression.Keywords
ERK5; P38; TNFΑ; VCAM-1; Hyperinsulinemia; AtherosclerosisIntroduction
Atherosclerosis is a major consequence of vascular dysfunction and in part leads to vascular smooth cell proliferation, lack of vascular compliance, endothelial cell remodeling, and vascular inflammatory events. One particular characteristic of endothelial pathology is the increased expression of cellular adhesion molecules (CAMs) at the surface of endothelial cells, which are located at the margins of the intima of the vessels [1-3].Although insulin is considered to be anti-atherogenic [4] other studies have indicated that long-term (i.e., chronic) insulin resistance accompanied by hyperinsulinemia contributes to the pathogenesis of atherosclerosis. The prolonged effects of insulin resistance are exhibited in part by the presence of inflammatory cytokines, which in turn significantly increase the expression of CAMs [5-10]. Tumor Necrosis Factor-alpha (TNFα) is one such cytokine. TNFα is secreted by mature macrophages during the progression of atherosclerosis, and is linked to insulin resistance and the production of cell surface molecules on endothelial cells such as Vascular Cellular Adhesion Molecule (VCAM-1) [11-13].
The intracellular effects of insulin and TNFα bring us to the question: which intracellular events that are associated with atherosclerosis are mediated by the major intracellular kinase pathways such as extracellular signal-regulated kinases-1/2 (ERK- 1/2), c-jun N-terminal kinases (JNK), p38 kinase, ERK5 and the canonical metabolic pathway, phosphatidylinositol-3 kinase (PI3K). Specifically, which ones of these kinases mediate insulin and TNFα− stimulated increases or decreases in expression of VCAM-1 is a question in this study.
We have previously measured the changes in VCAM-1 expression in the presence of insulin and TNFα. Additionally, we have previously observed insulin and TNFα stimulated VCAM-1 expression in the presence of inhibitory RNA of ERK2 and Akt. In this study, we wanted to determine the effects of insulin and TNFα in the presence of RNA interference (RNAi) with respect to ERK5 and p38. Here we report that (1) insulin and TNFα individually stimulated total and surface protein expression of VCAM-1 in Rat Aorta Vascular Endothelial Cells (RAVEC) greater than that seen in negative controls, (2) expression of RNAi ERK5 or p38 alone caused a significant (P < 0.05) decrease in insulin or TNFα-stimulated VCAM- 1 expression as compared to control cells, and (3) expression of RNAi ERK5 followed by expression of RNAi p38 caused a significant (P < 0.05) decrease of insulin and TNFα-stimulated VCAM-1 expression greater than that seen with RNAi of either alone.
Methods
MaterialsAll general lab reagents were purchased from Sigma-Aldrich (St. Louis, MO). Primary rabbit antibodies to ERK5 (3372S), p38 (9212), and alpha-tubulin (2144S) were from Cell Signaling (Boston, MA). The primary rabbit antibody to VCAM-1 (NBP1-95622) was from Novus Biologicals (Littleton, CO) and goat anti-rabbit-secondary antibody IRDye 680RD (926-68171) was from LI-COR (Lincoln, NE). DyLight 488-conjugated anti-VCAM-1 antibody was from Thermo Fisher Scientific (Pittsburgh, PA). Four-well chamber slides were from Thermo Fisher and DAPI Mounting Medium was from Vector Labs (Burlingame, CA). PVDF membranes and other Western blot accessories were from GE Healthcare/Amersham (Piscataway, NJ). Rat aorta vascular endothelial cells (RAEC) (CRL-1444) were from ATCC (Manassas, VA) and culture medium was from Life Technologies (Grand Island, NY).
Transfection medium (sc-108062) and transfection reagent (sc- 108061) were from Santa Cruz (Dallas, TX) ERK5 shRNA (K447075P, 336314) and p38 shRNA (KR52703P, 336314) were obtained from SA Biosciences/Qiagen (ThermoFisher) Valencia, CA.
Cell culturing
RAEC were cultured in complete growth medium (CGM) [DMEM with 4 mM L- glutamine, 4.5 g/L-glucose and 1.5 g/Lsodium bicarbonate) and supplemented with 10% heat- inactivated fetal bovine serum (HI-FBS) (10438-026) (Life Technologies, Grand Island, NY) and 1% Antimycotic-Antibiotic solution (15240-062)(Life Technologies) and cultured at 37 °C, 5% CO2 atmosphere.
Preparation of shRNA stable cell lines
RAEC were grown to 50-70% confluence in CGM in 6-well culture plates. Cells were transfected with either shERK5 inhibitory plasmids 1-4, shp38 inhibitory plasmids 1-4, or control scrambled plasmids as previously described [14]. RNAi was delivered to cells via a transfection solution (medium and reagent) as per vendor’s protocol. Mock transfections were completed by incubating cells in the transfection solution minus any RNAi and were designated as “vehicle”. Cells were incubated in CGM containing 2 μg/mL of Puromycin (Sigma-Aldrich) for 2-3 weeks for selection of Puromycin resistant transformants.
Dual transfection of stable cell lines
To examine the effect of simultaneous ERK5 and p38 knockdown on VCAM-1 expression, the ERK5 shRNA stable cell line (ERK5 KD) was transiently transfected with shp38 plasmid and the p38 shRNA stable cell line (p38 KD) was transiently transfected with the shERK5 plasmid. These two protocols were carried out in order to see if any difference occurred with respect to transfection sequence. Stable cell lines were transiently transfected with shRNA plasmid DNA as described above and incubated for 5 hr with the DNA transfection mix. Subsequently the transfection mix was aspirated and replaced with 2.0 mL CGM. Stimulation of cells by insulin and/or TNFα occurred 48 hours after transient transfection was accomplished.
Stimulation of VCAM-1 expression
RAEC were cultured in CGM, whereas shRNA stable cell lines (e.g. ERK2 KD and p38 KD) were cultured in CGM containing 2 μg/ mL Puromycin until assays were performed. After incubating the transfected cells for an additional 48 hr, the cells were stimulated with or without TNFα (10 ng/mL) and in the presence or absence of insulin (10 nM). Doses of TNFα and insulin have been previously determined using dose-response and time-course curves.
Thereafter, cells were evaluated for VCAM-1 expression as previously described [14]. Briefly, TNFα stimulation occurred over a total of 6 hr, within such time the cells were stimulated with insulin for 1 hr.
Western blot analysis
Sodium Dodecyl Sulfate Polyacrylamide Electrophoresis was performed on cleared lysates. Western blot analysis was subsequently performed as previously described [14], with the following differences. After completion of protein transfer, membranes were washed in ultra-pure water for 5 min. Membranes were then incubated in 3% non-fat milk (milk) in Tris- buffered Saline (TBS) blocking solution for 1 hr at room temperature and then incubated with a designated primary antibody solution (1:1000 in 3% milk/TBS-T) overnight at 4 °C. Membranes were washed 4 times with TBS plus Tween (TBS-T) for 5 min at room temperature and then incubated with a goat anti-rabbit secondary antibody (1:5000 in 3% milk/ TBS-T) conjugated to fluorochrome IR680RD for 1 hour at room temperature. Membranes were washed 4 times with TBS-T for 5 min each time at room temperature and then incubated with a rabbit antitubulin primary antibody solution (1:1000 in 3% milk/TBS-T) for 3 hr at room temperature. After washing the membranes for 4 times with TBS-T, the membranes were again incubated with a goat antirabbit secondary antibody (1:5000 in 3% milk/TBS-T) conjugated to fluorochrome IR680RD for 1 hour at room temperature. The membranes were washed 4 times with TBS-T and allowed to dry before performing densitometry. Densitometry was performed using an Odyssey Licor system (Lincoln, NE). Alpha-tubulin or actin protein was used to normalize VCAM-1 signals.
Flow cytometry
Non-transfected RAEC was controls. Stably transfected RAEC ERK5 knockdown (ERK5 KD) and p38 knockdown (p38 KD) cell lines were inoculated into 6-well tissue culture dishes, transiently transfected with vehicle or shp38 or shERK5 plasmids, respectively, and stimulated with insulin, TNFα, or combined insulin and TNFα as described above. The cells were washed twice with 2 mL of 1X PBS (Gibco). The PBS was aspirated and 0.5 mL of Cell Dissociation Solution Non-Enzymatic (Sigma-Aldrich) was added to each well. After incubating the cells at 37 °C and 5% CO2 for 30 min, 1 mL of 1% Bovine Serum Albumin (BSA, Sigma-Aldrich) in PBS was added to the cells and then were gently triturated into a single cell suspension. The cells were transferred to 5 mL Falcon polystyrene round bottom tubes (Thermo Scientific) and centrifuged at 500 x g for 5 min. After aspirating the supernatants, the cells were resuspended in mL 1% BSA, pelleted at 500 x g by centrifugation, and the supernatants removed by aspiration. The cells were resuspended in 200 μL of 1% BSA. Two microliters of DyLight 488- conjugated anti-VCAM-1 antibody (Life Technologies, Grand Island, New York) were added to each tube and the cells were resuspended by vortexing. The cells were incubated in the dark for 30 min at room temperature. The cells were centrifuged, washed twice with 3 mL 1% BSA and resuspended in 200 μL of 1% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA). After incubating the cells for 5 min at room temperature, the cells were diluted with an additional 300 μL of PBS and analyzed using flow cytometry. The experiments were run on a BD LSRII (BD Biosciences, San Jose, CA). MFI and gating percentages as part of data analysis was done using BD FACS Diva v6 software.
Chamber slide cell preparation
2 x 105 of control untransfected RAEC, ERK5 KD or p38 KD stable cell lines were plated in 1 mL of CGM in each well of a 4-well chamber slide and allowed to grow for 24 h, 37 °C and 5% CO2. The medium was aspirated and 1.0 mL of fresh CGM was applied to the cells. ERK5 KD and p38 KD were either mock transfected, or transiently transfected with shp38 or shERK5, respectively for 48 hours. Cells were then treated with either vehicle [Santa Cruz’s transfection reagent and medium combination (as per vendor’s DNA transfection protocol)], 10 ng/mL TNFα or insulin (10 nM) alone or in combination. The medium was aspirated and the cells were washed three times with PBS and then incubated in 400 μL of 4% paraformaldehyde in PBS for 30 minutes at room temperature. The medium was aspirated and washed three times with 1 mL of PBS. The final PBS wash was aspirated and 400 μL of a 1:1000 DyLight anti- VCAM-1 antibody solution in 1% BSA was added to each chamber and incubated for 30 minutes at room temperature. The cells were then washed three times with 500 μL of 1% BSA. The chamber walls were removed and one drop of DAPI Mounting Medium was added to each group of cells on the slide. Cells were then sealed with a glass cover slip using clear nail polish. Slides were kept in a dark refrigerator until microscopic visualization.
Confocal microscopy
Cells were imaged with a Leica TSC SP8X white light laser scanning confocal microscope (Leica Microsystems GmbH. Ernst-Leitz-Straße 17-37 Wetzlar, 35578 Germany). All image acquisitions were carried out using the Leica Application Suite X (version 1.1.0.12420, LASX AF). Excitation of the DAPI channel was accomplished using a 405 nm diode laser with an excitation intensity level of 2.67%. Emission signal was captured with standard PMT Channel 1 and an emission gap of 430 nm-480 nm. The Leica Supercontinuum white light excitation laser line (488nm) with 3% intensity level was used to for Alexa fluor 488. Emission signals were captured with the Leica HyD 2 detector (Hybird 2 PMT) with an emission gap of 505 nm-555 nm.
Data analysis
Data were analyzed by either unpaired Student’s t test (two groups) or ANOVA with subsequent Tukey post hoc test (several groups) as indicated. A “P” value of less than 0.05 was considered significant. Results were expressed as the Mean ± Standard Error of the Mean (SEM) of three or more independent experiments.
Results
Since we previously investigated the intracellular expression of total VCAM-1 protein in the presence of insulin and/or TNFα [15], we extended our studies by examining the changes in expression of VCAM-1 both in total protein and at the cell surface in the presence of knockdown ERK5 (ERK5 KD) and p38 cell lines (p38 KD).In order to measure changes in VCAM-1 expression in ERK5 KD and p38 KD cells, we first needed to determine whether or not cells transfected with shERK5 and shp38 plasmid constructs could significantly decrease ERK5 and p38 protein, respectively. Using short hairpin (sh) plasmid clones, we observed that clone #1 of shERK5 and clone #1 of shp38 significantly (P < 0.05) decreased their respective endogenous proteins 38% and 27%, respectively (Figure 1). Using the most effective clone for each kinase, we then transfected RAEC with the appropriate clones and then treated the cells with insulin (10 nM, 1 h) or TNFα (10 ng/mL, 6 h) alone or in combination and thereafter measured changes in total VCAM-1 protein using Western blot analyses (Figure 2). In order to verify that transient transfection of shp38 decreased p38 protein in ERK KD cells, we transfected stable ERK5 KD cells with shp38. Amounts of ERK5 and p38 were analyzed at 48 hours after shp38 transfection (Figure 2A). In cells treated with insulin alone, we observed that ERK5 KD and ERK5 KD plus shp38 significantly (P < 0.05) decreased total VCAM-1 protein as compared to insulin-stimulated positive controls (Figure 2B). In contrast, cells treated with TNFα alone and transfected with either ERK5 KD alone or ERK5 KD plus shp38 exhibited a decrease of 43% and 68%, respectively, compared to positive controls (cells not transfected with shRNA) (Figure 2C). Likewise, ERK5 KD and ERK5 KD plus shp38 cells treated with insulin plus TNFα exhibited a decrease of 30% and 50%, respectively, in VCAM-1 protein as compared to positive, nontransfected controls.
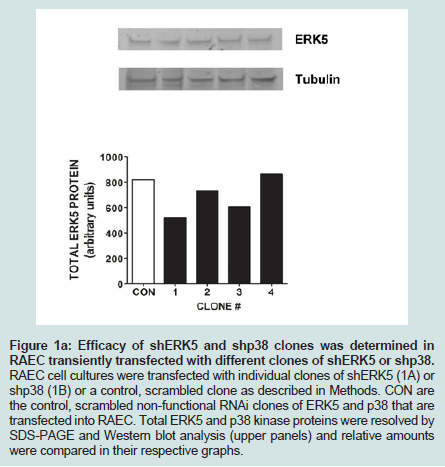
Figure 1A: Efficacy of shERK5 and shp38 clones was determined in RAEC transiently transfected with different clones of shERK5 or shp38. RAEC cell cultures were transfected with individual clones of shERK5 (1A) or shp38 (1B) or a control, scrambled clone as described in Methods. CON are the control, scrambled non-functional RNAi clones of ERK5 and p38 that are transfected into RAEC. Total ERK5 and p38 kinase proteins were resolved by SDS-PAGE and Western blot analysis (upper panels) and relative amounts were compared in their respective graphs..
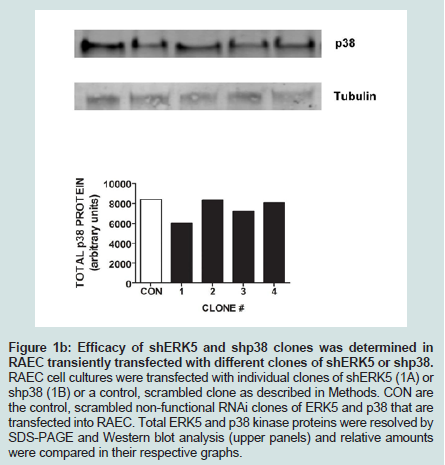
Figure 1B: Efficacy of shERK5 and shp38 clones was determined in RAEC transiently transfected with different clones of shERK5 or shp38. RAEC cell cultures were transfected with individual clones of shERK5 (1A) or shp38 (1B) or a control, scrambled clone as described in Methods. CON are the control, scrambled non-functional RNAi clones of ERK5 and p38 that are transfected into RAEC. Total ERK5 and p38 kinase proteins were resolved by SDS-PAGE and Western blot analysis (upper panels) and relative amounts were compared in their respective graphs.
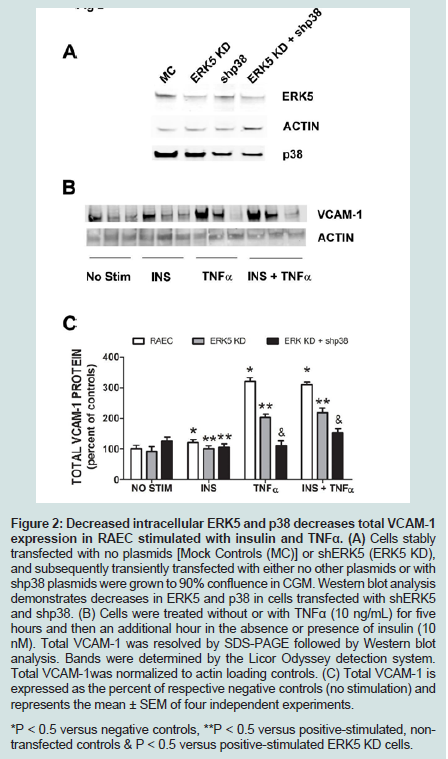
Figure 2: Decreased intracellular ERK5 and p38 decreases total VCAM-1 expression in RAEC stimulated with insulin and TNFα. (A) Cells stably transfected with no plasmids [Mock Controls (MC)] or shERK5 (ERK5 KD), and subsequently transiently transfected with either no other plasmids or with shp38 plasmids were grown to 90% confluence in CGM. Western blot analysis demonstrates decreases in ERK5 and p38 in cells transfected with shERK5 and shp38. (B) Cells were treated without or with TNFα (10 ng/mL) for five hours and then an additional hour in the absence or presence of insulin (10 nM). Total VCAM-1 was resolved by SDS-PAGE followed by Western blot analysis. Bands were determined by the Licor Odyssey detection system. Total VCAM-1was normalized to actin loading controls. (C) Total VCAM-1 is expressed as the percent of respective negative controls (no stimulation) and represents the mean ± SEM of four independent experiments. *P < 0.5 versus negative controls, **P < 0.5 versus positive-stimulated, nontransfected controls & P < 0.5 versus positive-stimulated ERK5 KD cells.
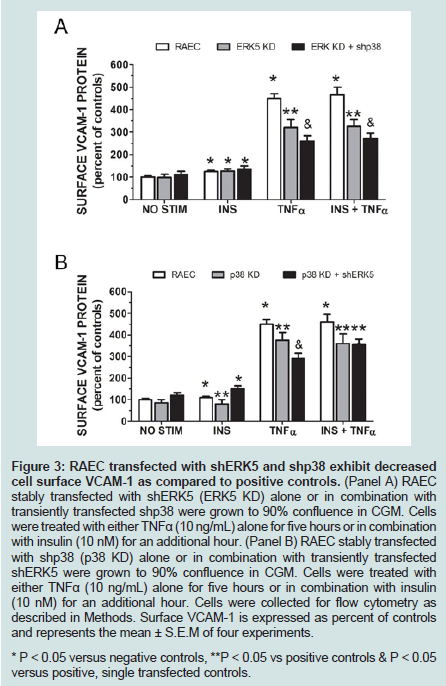
Figure 3: RAEC transfected with shERK5 and shp38 exhibit decreased cell surface VCAM-1 as compared to positive controls. (Panel A) RAEC stably transfected with shERK5 (ERK5 KD) alone or in combination withtransiently transfected shp38 were grown to 90% confluence in CGM. Cells were treated with either TNFα (10 ng/mL) alone for five hours or in combination with insulin (10 nM) for an additional hour. (Panel B) RAEC stably transfected with shp38 (p38 KD) alone or in combination with transiently transfected shERK5 were grown to 90% confluence in CGM. Cells were treated with either TNFα (10 ng/mL) alone for five hours or in combination with insulin (10 nM) for an additional hour. Cells were collected for flow cytometry as described in Methods. Surface VCAM-1 is expressed as percent of controls and represents the mean ± S.E.M of four experiments. * P < 0.05 versus negative controls, **P < 0.05 vs positive controls & P < 0.05 versus positive, single transfected controls.
Finally, to corroborate the changes in VCAM-1 expression observed in flow cytometry studies, we performed confocal microscopy (Figure 4). ERK5 KD or p38 KD transfected cells, or the combination (i.e. ERK5 KD plus shp38 or p38 KD plus shERK5) exhibited decreased VCAM-1 expression at the cell surface compared to positive control cells, thus supporting the experiments of flow cytometry.
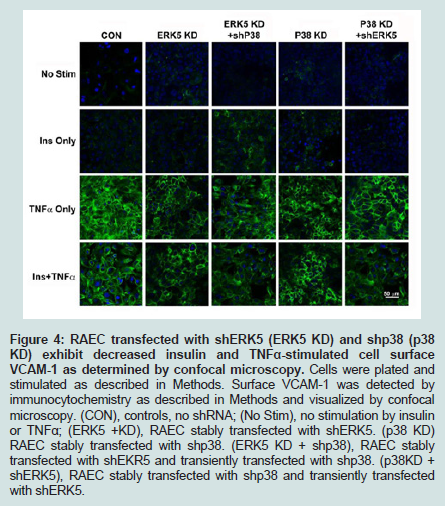
Figure 4: RAEC transfected with shERK5 (ERK5 KD) and shp38 (p38 KD) exhibit decreased insulin and TNFα-stimulated cell surface VCAM-1 as determined by confocal microscopy. Cells were plated and stimulated as described in Methods. Surface VCAM-1 was detected by immunocytochemistry as described in Methods and visualized by confocal microscopy. (CON), controls, no shRNA; (No Stim), no stimulation by insulin or TNFα; (ERK5 +KD), RAEC stably transfected with shERK5. (p38 KD) RAEC stably transfected with shp38. (ERK5 KD + shp38), RAEC stablytransfected with shEKR5 and transiently transfected with shp38. (p38KD + shERK5), RAEC stably transfected with shp38 and transiently transfected with shERK5.
Discussion
Type-2 Diabetes Mellitus (T2DM) is a syndrome of multiple disorders that starts with insulin resistance and eventually leads to a multitude of pathologies. Vascular complications are the most prevalent pathology found associated with T2DM. Insulin resistance and hyperinsulinemia are risk factors for cardiovascular disease (CVD) [16,17], which in time and unrestricted will lead to morbidity and mortality. Inflammation of the vasculature is associated with T2DM and the expression of cellular adhesion molecules such as VCAM-1, which is upregulated in the state of insulin resistance and hyperinsulinemia [15] leads to the exacerbation of this syndrome.Although the serum milieu, which is external to the endothelial cell is important to the overall condition of the blood, blood-borne hormones and cytokines have profound effects on the internal events of vascular cells. When these external signals are transduced into the cells via cell surface receptors and these signals are then conveyed from cell membrane to internal cellular targets via kinase pathways. We report here that ERK5 and p38 appear to be important mediators of insulin and TNFα-stimulated VCAM-1 expression. We show here that when ERK5 or p38 kinase is downregulated in rat aorta endothelial cells via RNAi interference, there is a significant (P < 0.05) decrease in insulin and TNFα stimulated total and surface VCAM-1 expression. Additionally, when both ERK5 and p38 kinases are downregulated, there is even a greater and significant (P < 0.05) decrease in VCAM-1 protein both at the cell surface and in the cytoplasm.
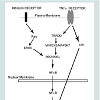
The putative mechanism for this phenomenon may lie in three loci of the cell: (1) the receptors for insulin and TNFα, (2) their respective proximal kinase mediators and (3) common junctions downstream of the upstream mediators. There appears to be no promiscuity between insulin and TNFα for the other’s receptor. Thus, this establishes a scenario for an additive effect of these two pathways on downstream events. The proximal effector molecule of the insulin receptor is Ras, whereas Tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD) is the effector molecule for the TNFα receptor. Yet, downstream of these adaptor proteins are their proximal kinases of the MAP kinase superfamily. We suggest that the kinases involved in this scenario are ERK5 for the insulin receptor and Mitogen - Activated Protein 2-Kinase-3 (MAP2K3) and MAP3K7 for the TNFα receptor. The convergence point of these distal and diverse kinases pathways is the canonical inflammatory intermediate Nuclear Factor kappa-B (NFκB). Parallel to these kinase signal pathways is the MAP2K4-mediated pathway which converges with others to its target, p38 kinase. Both activated NFκB and p38 kinase translocate from the cytoplasm to the nucleus and bind there cognate response elements, upregulating gene expression.
What is surprising in this study is the lack of potentiation of insulin on TNFα-stimulated VCAM-1 expression (Figures 2 and 3). One could speculate that with two different pathways and upstream effectors of NFκB that signaling from one receptor would augment that seen from the other. Yet, this was not demonstrated in this study. Two possible explanations may be proffered here: (1) the concentrations of insulin and TNFα in these experiments are superphysiological and therefore no potentiation can occur, or (2) the length of time for insulin and TNFα to stimulate the cells did not mimic patho- physiologic parameters; that is, long-term chronic conditions. Future studies are forthcoming in order to tease out these anomalies. Different combination of doses and longer time points may be necessary to reach the expected potentiation of insulin on TNFα-stimulated VCAM-1 expression.
Inflammation of the vasculature is associated with T2DM and the expression of cellular adhesion molecules on the surface of vascular endothelial cells. VCAM-1 is one such adhesion molecule. TNFα is a well characterized inflammatory cytokine and its presence is robustly correlated with atherogenesis [7,18]. In contrast, insulin’s role in atherogenesis is hotly debated. Some contend that insulin stimulates an increase in nitric oxide (NO) and a decrease in systemic inflammation [4,19], both of which appear to inhibit atherogenesis. Others argue that insulin, especially hyperinsulinemia, increases the production of endothelial and vascular smooth muscle cells, adding to the development of atherosclerotic plaques [20,21].
Previous studies have demonstrated that (1) hyperinsulinemia is positively associated with atherosclerosis [22-24], (2) CAM expression is linked to inflammatory conditions and atherosclerosis [25,26] and (3) insulin resistance and hyperinsulinemia contribute to increased expression of TNFα with subsequent increased expression of VCAM-1 [27].
Yet, the two sides are not incompatible since insulin appears to have multiple effects. At physiologic concentrations, insulin has a vaso-protective effect [4]. However, at higher than physiologic concentrations, chronic hyperinsulinemia appears to have a detrimental effect by enhancing the effects of deleterious cytokines. In addition, previous reports demonstrate that high physiologic insulin levels may inhibit endothelial progenitor cell proliferation [28,29].
Finally, others have demonstrated that hyperphysiologic insulin appears to augment more potent growth factors such as plateletderived growth factor [11,30], thus contributing to destructive effects on the endothelium.
Although expression of VCAM-1 and its cousin Interstitial Adhesion Molecule-1 (ICAM-1) appear to increase in expression within the context of insulin resistance and hyperinsulinemia, VCAM-1 may play a more dominant role in atherosclerosis than ICAM-1 [12]. One possible mechanism for increased expression of CAMs is based on the observation that insulin may prepare cells to be more responsive to potent cytokines, which in turn complements the insulin resistant state and thereby increases more CAM expression [31].
We have shown in this study that high-physiological concentrations of insulin and TNFα increase the expression of VCAM-1 in RAEC. In contrast, RAEC stably transfected with shERK5 and shp38, exhibit decreased amounts of VCAM-1 expression compared to positive controls. These results suggest that ERK5 and p38 are positive regulators of VCAM-1 expression and their decreased expression results in decreased total and surface VCAM-1 expression (Figure 5).
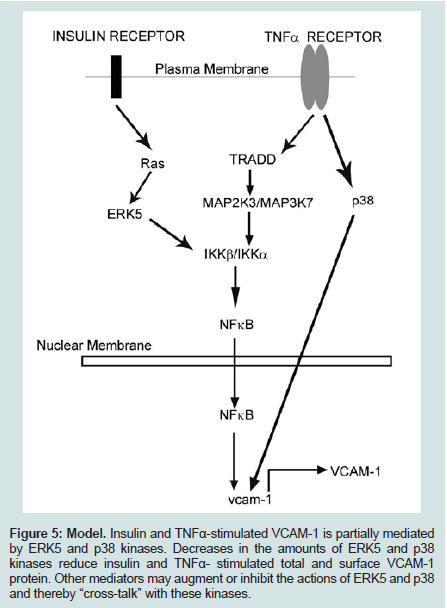
Figure 5: Model. Insulin and TNFα-stimulated VCAM-1 is partially mediated by ERK5 and p38 kinases. Decreases in the amounts of ERK5 and p38 kinases reduce insulin and TNFα- stimulated total and surface VCAM-1 protein. Other mediators may augment or inhibit the actions of ERK5 and p38 and thereby “cross-talk” with these kinases.
Acknowledgements
We would also like to acknowledge the assistance of Harsh Pratap (Flow Cytometry technician for the Mucosal and Vaccine Research Colorado) and Ron Bouchard (Microscopy Core), both of the Eastern Colorado Health Care Service (Denver VAMC).FundingThis work was supported by the Research Service of the Department of Veterans Affairs (to M.L.G.), in which Dr. Goalstone is a recipient of a VA Career Development Award.References
- Madonna R, Pandolfi A, Massaro M, Consoli A, De Caterina R (2004) Insulin enhances vascular cell adhesion molecule-1 expression in human cultured endothelial cells through a pro-atherogenic pathway mediated by p38 mitogen-activated protein-kinase. Diabetologia 47: 532-536.
- Okouchi M, Okayama N, Imai S, Omi H, Shimizu M, et al. (2002) High insulin enhances neutrophil transendothelial migration through increasing surface expression of platelet endothelial cell adhesion molecule-1 via activation of mitogen activated protein kinase. Diabetologia 45: 1449-1456.
- Watson PA, Nesterova A, Burant CF, Klemm DJ, Reusch JE (2001) Diabetes-related changes in cAMP response element-binding protein content enhance smooth muscle cell proliferation and migration. J Biol Chem 276: 46142-46150.<
- Aljada A, Saadeh R, Assian E, Ghanim H, Dandona P (2000) Insulin inhibits the expression of intercellular adhesion molecule-1 by human aortic endothelial cells through stimulation of nitric oxide. J Clin Endocrinol Metab 85: 2572-2575.
- Lele RD (2007) Causation, prevention and reversal of vascular endothelial dysfunction. J Assoc Physicians India 55: 643-651.
- Goldschmidt-Clermont PJ, Creager MA, Lorsordo DW, Lam GK, Wassef M, et al. (2005) Atherosclerosis 2005: recent discoveries and novel hypotheses. Circulation 112: 3348-3353.
- Dessein PH, Joffe BI, Singh S (2005) Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res Ther 7: R634-R643.
- Endemann DH, Schiffrin EL (2004) Endothelial dysfunction. J Am Soc Nephrol 15: 1983-1992.
- Huo Y, Ley K (2001) Adhesion molecules and atherogenesis. Acta Physio Scand 173: 35-43.
- Ginsberg HN (2000) Insulin resistance and cardiovascular disease. J Clin Invest 106: 453-458.
- Yamaguchi K, Higashiura K, Ura N, Murakami H, Hyakukoku M, et al. (2003) The effect of tumor necrosis factor-alpha on tissue specificity and selectivity to insulin signaling. Hypertens Res 26: 389-396.
- Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, et al. (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 107: 1255-1262.
- Barks JL, McQuillan JJ, Iademarco MF (1997) TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol 159: 4532-4538.
- Mackesy DZ, Goalstone ML (2014) Extracellular signal-regulated kinase-5: Novel mediator of insulin and tumor necrosis factor α-stimulated vascular cell adhesion molecule-1 expression in vascular cells. J Diabetes 6: 595-602.
- Mackesy DZ, Goalstone ML (2011) Insulin augments tumor necrosis factor-alpha stimulated expression of vascular cell adhesion molecule-1 in vascular endothelial cells. J Inflamm (Lond) 8: 34
- Lebovitz HE (1999) Type 2 diabetes: an overview. Clin Chem 45(8 Pt 2): 1339-1345.
- Lu H, Raptis M, Black E, Stan M, Amar S, et al. (2004) Influence of diabetes on the exacerbation of an inflammatory response in cardiovascular tissue. Endocrinology 145: 4934-4939.
- Narizhneva NV, Razorenova OV, Podrez EA, Chen J, Chandrasekharan UM, et al. (2005) Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. FASEB J 19: 1158-1160.
- Breen DM, Chan KK, Dhaliwall JK, Ward MR, Al Koudsi N, et al. (2009) Insulin increases reendothelialization and inhibits cell migration and neointimal growth after arterial injury. Arterioscler Thromb Vasc Biol 29: 1060-1066.
- Wang CC, Sharma G, Draznin B (2006) Early growth response gene-1 expression in vascular smooth muscle cells effects of insulin and oxidant stress. Am J Hypertens 19: 366-372.
- Golovchenko I, Goalstone ML, Watson P, Brownlee M, Draznin B (2000) Hyperinsulinemia enhances transcriptional activity of nuclear factor-kappaB induced by angiotensin II, hyperglycemia and advanced glycosylation end products in vascular smooth muscle cells. Circ Res 87: 746-752.
- Festa A, D'Agostino R Jr, Mykkanen L, Tracy RP, Zaccaro DJ, et al. (1999) Relative contribution of insulin and its precursors to fibrinogen and PAI-1 in a large population with different states of glucose tolerance. The Insulin Resistance Atherosclerosis Study (IRAS). Arterioscler Thromb Vasc Biol 19: 562-568.
- Kekalainen P, Sarlund H, Farin P, Kaukanen E, Yang X, et al. (1996) Femoral atherosclerosis in middle-aged subjects: association with cardiovascular risk factors and insulin resistance. Am J Epidemiol 144: 742-748.
- Standl E (1995) Hyperinsulinemia and atherosclerosis. Clin Invest Med 18: 261-266.
- van de Stolpe A, van der Saag PT (1996) Intercellular adhesion molecule-1. J Mol Med (Berl) 74: 13-33.
- Watanabe T, Fan J (1998) Atherosclerosis and inflammation mononuclear cell recruitment and adhesion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. Int J Cardiol 66 Suppl 1: S45-S53.
- Dandona P, Weinstock R, Thusu K, Abdel-Rahman E, Aljada A, et al. (1998) Tumor necrosis factor-alpha in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 83: 2907-2910.
- Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA (2000) Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest 106: 571-578.
- Zhang W, Wang X, Jin H, Qian R, Zhang G, et al. (2008) Effects of high glucose plus high insulin on proliferation and apoptosis of mouse endothelial progenitor cells. Inflamm Res 57: 571-576.
- Goalstone ML, Natarajan R, Standley PR, Walsh MF, Leitner JW, et al. (1998) Insulin potentiates platelet-derived growth factor action in vascular smooth muscle cells. Endocrinology 139: 4067-4072.
- Goalstone ML, Leitner JW, Draznin B (1999) Mechanism of insulin's ability to potentiate mitogenic effects of growth factors, 1999. Ad Mol Cell 3: 155-173.