
Journal of Pharmaceutics & Pharmacology
Download PDF
Research Article
 The metabolizing enzyme activity of a drug is often modulated by the co-administration of inhibitors or inducers that affect the drug-metabolizing enzyme. Therefore, the prediction of drug-drug interactions related to the drug metabolism in humans is typically examined in an invitro metabolism study of tissue sub cellular fractions or in invivo pharmacokinetic studies using experimental animals. The most important enzyme in drug metabolism, cytochrome P450s (CYPs), is involved in metabolic interactions with other drugs. MP has been used since a long time ago as an CYP inhibitor in invitro and invivo studies in the rats [13,14]. As described above, although it was suggested that 11β-HSD1 contributes to the reductive metabolism of MP to MPOL, in so far we know, there is no report of drug-drug interactions towards the metabolism of MP by an 11β-HSD1 inhibitor. Therefore, it is not clear whether the inhibitor affects the pharmacokinetics of MP and its metabolites. Of the known 11β-HSD1 inhibiters [15,16], we focused our attention on glycyrrhetinic acid (GA) and examined its effect on the metabolism of MP in the rat liver and testis invitro and the pharmacokinetics invivo. The finding reported here in provide new evidence to show that the use of GA as an inhibitor of 11β-HSD1is related to the reduction of MP to MPOL in the microsomal and mitochondrial fractions of the testis as well as the liver in Wistar male rats. In addition, we show the first time, that the pharmacokinetics of MP and its metabolites are modulated by the co-administration of GA to the rats.
The metabolizing enzyme activity of a drug is often modulated by the co-administration of inhibitors or inducers that affect the drug-metabolizing enzyme. Therefore, the prediction of drug-drug interactions related to the drug metabolism in humans is typically examined in an invitro metabolism study of tissue sub cellular fractions or in invivo pharmacokinetic studies using experimental animals. The most important enzyme in drug metabolism, cytochrome P450s (CYPs), is involved in metabolic interactions with other drugs. MP has been used since a long time ago as an CYP inhibitor in invitro and invivo studies in the rats [13,14]. As described above, although it was suggested that 11β-HSD1 contributes to the reductive metabolism of MP to MPOL, in so far we know, there is no report of drug-drug interactions towards the metabolism of MP by an 11β-HSD1 inhibitor. Therefore, it is not clear whether the inhibitor affects the pharmacokinetics of MP and its metabolites. Of the known 11β-HSD1 inhibiters [15,16], we focused our attention on glycyrrhetinic acid (GA) and examined its effect on the metabolism of MP in the rat liver and testis invitro and the pharmacokinetics invivo. The finding reported here in provide new evidence to show that the use of GA as an inhibitor of 11β-HSD1is related to the reduction of MP to MPOL in the microsomal and mitochondrial fractions of the testis as well as the liver in Wistar male rats. In addition, we show the first time, that the pharmacokinetics of MP and its metabolites are modulated by the co-administration of GA to the rats.
Invitro and Invivo Inhibition of Metyrapone metabolism by Glycyrrhetinic Acid in Male Wistar Rats
Hideyuki Murata1,2, Toshiyuki Higuchi2* and Masaki Otagiri3
- Beijing Sciecure Pharmaceutical Co., Ltd., Industrial Park, Shunyi District, Beijing, China
*Address for Correspondence: Toshiyuki Higuchi, Department of Health Biosciences, Nihon Pharmaceutical University, 10281 Komuro, Ina-machi, Kitaadachigun, Saitama 362-0806, Japan, Tel: +81-48-721-1155; Fax: +81-48-721-6718; E-mail: higuchi@nichiyaku.ac.jp
Citation: Murata H, Higuchi T, Otagiri M. Invitro and Invivo Inhibition of Metyraponemetabolism by Glycyrrhetinic Acid in Male Wistar Rats. J Pharmaceu Pharmacol. 2016;4(1): 6.
Copyright & copy; © 2016 Murata H et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology | Volume: 4, Issue: 1
Submission: 01 November, 2016 | Accepted: 12 December, 2016 | Published: 19 December, 2016
Citation: Murata H, Higuchi T, Otagiri M. Invitro and Invivo Inhibition of Metyraponemetabolism by Glycyrrhetinic Acid in Male Wistar Rats. J Pharmaceu Pharmacol. 2016;4(1): 6.
Copyright & copy; © 2016 Murata H et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology | Volume: 4, Issue: 1
Submission: 01 November, 2016 | Accepted: 12 December, 2016 | Published: 19 December, 2016
Abstract
Objectives: We investigated the inhibitory effect of glycyrrhetinic acid (GA), an 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor, on the reductive metabolism of metyrapone (MP) in an invitro study, and the effect of the co-administration of GA on the pharmacokinetics of MP and its metabolites in rats.
Methods: High-performance liquid chromatography was used to determine the metabolic activities of MP in the liver and testis and the plasma concentrations of MP and its metabolites after MP with or without a GA dose to rats.
Results: GA strongly inhibited the reduction of MP to metyrapol(MPOL) in the microsomal and mitochondrial fractions of both the tissues in a dose-dependent manner. When GA was co-administrated, the plasma levels of MPOL and MPOL N-oxide II (MPOL NOII) were lower than those of MP alone, while the levels of MP and MP NOII were higher than those of MP alone.
Conclusion: The in vitro GA inhibition suggests that the reduction of MP in the rat liver and testis is catalyzed by 11β-HSD1. The coadministration of GA modulated the plasma pharmacokinetics of MP, MPOL, MP NOII and MPOL NOII, suggesting that the changes in the pharmacokinetics were caused by the inhibition of 11β-HSD1 invivo.
Keywords
Metyrapone; 11β-hydroxysteroid dehydrogenase 1; Male rats; Liver; Testis; Invivo drug-drug interaction
Introduction
11β-hydroxysteroid dehydrogenase (11β-HSD) belongs to the short-chain dehydrogenase/reductase super family, and plays an important role in the reduction of ketone groups of various drugs [1-3]. Two isozymes of 11β-HSD, 11β-HSD type1 (11β-HSD1) and 11β-HSD type2 (11β-HSD2), are known, and distributed in a variety of tissues [3,4]. For 11β-HSD1, high expression levels are observed in the liver, reproductive organs and adipose tissue, with high levels of 11β-HSD2 in the kidney and colon [5,6]. Overall, the enzymes are located in the microsomal fraction of the tissues, in which 11β-HSD1 and 11β-HSD2 have NADP(H)-dependent glucocorticoid 11-oxidoreductase activity and NAD+-dependent glucocorticoid 11-hydroxydehydrogenase activity, respectively [1,6,7]. It has been reported that a higher expression of 11β-HSD1in the adipose tissue increases endogenous cortisol concentrations, resulting in the development of Cushing’s syndrome [8,9].
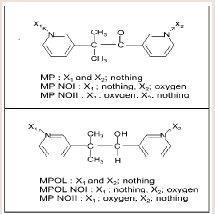
Metyrapone (MP) (Figure 1) is employed as a therapeutic drug for the treatment of Cushing’s syndrome, because the drug suppresses cortisol biosynthesis by inhibiting 11β-hydroxylase activity [10]. MP which contains a ketone group is reduced to a secondary alcohol metabolite, metyrapol (MPOL) (Figure 1). Our recent study [11] demonstrated that, after a single oral administration of MP to Wistar male rats, MPOL was the major metabolite and its concentration was maintained at levels higher than these for MP in plasma. The major site involved in the reduction to MPOL in the rats is the liver, and the high MP reducing activity is presented in the liver microsomal fraction [11]. Additionally, we previously reported that the rat liver MPreducing enzyme is present in not only the microsomal fraction but also the mitochondrial fraction [11]. Since many drug-metabolizing enzymes including reductase activity for ketone-containing drugs are present in the mitochondrial fraction [12], we hypothesized that the rat mitochondrial fraction plays a role in the reduction of MP in addition to the microsomal fraction. Furthermore, we proposed that the major enzyme involved in reduction of MP to MPOL is 11β-HSD1 in both sub cellular fractions similar to the human liver [11]. Therefore, it is conceivable that the rats area relevant species for evaluating human subjects in predicting the involvement of 11β-HSD1in these metabolic steps. Interestingly, high MP reduction activity was found in both fractions derived from rat testis [11]. However, the enzyme involved in this conversion in this tissue is not well understood.
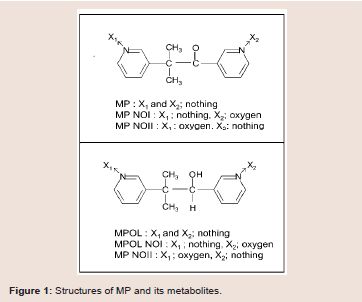
Figure 1: Relationship between the factors and responses [4].
Materials and Methods
Chemicals
MP and glycyrrhetinic acid (GA) were purchased from Sigma aldrich (Tokyo, Japan). Five metabolites of MP, MPOL, the form in which either of two pyridyl nitrogen atoms, MP N-oxide I (MP NOI) and MP N-oxide II (MP NOII), and the form in which the ketone group is reduced, MPOL N-oxide I (MPOL NOI) and MPOL N-oxide II (MPOL NOII) (Figure 1) were synthesized according to our previous report [11]. NADP+, glucose-6-phosphate (G-6-P) and glucose-6-phosphate dehydrogenase (G-6-PDH) were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan). Magnesium chloride 6H2O was purchased from Nacalai tesque (Tokyo, Japan). Highperformance liquid chromatography (HPLC) grade methanol and Milli-Q water were used to prepare the mobile phase for the HPLC analyses. All chemicals and solvents were commercial grades of highest purity or HPLC grade.
Animals
Male Wistar rats (9 weeks old, 200-240 g) were purchased from Japan SLC Inc., (Shizuoka, Japan). All animal experiments were approved by the Animal Experimentation Ethics Committee in Nihon Pharmaceutical University (permit numbers: 15-11; 24-September-2015, 15-15; 5-October-2013), and performed in accordance with the Guidelines for Animal Experimentation of Nihon Pharmaceutical University.
Preparation of subcellular fractions
Blood from rats which were not administered MP and GA was withdrawn any from the abdominal aorta. The livers perfused with ice-cold 1.15% KCl and the testis were isolated and homogenized in three volumes of homogenization buffer, 10 mM phosphate buffer (pH 7.4) containing 1.15% KCl using a glass-Teflon Potter Elvenhjem homogenizer (Iwaki Glass Co. Ltd., Tokyo, Japan). All subsequent procedures were performed at 4 °C. The mitochondrial and microsomal fractions were prepared based on our previous report [11].
MP-metabolizing enzyme assays
For the assay of MP reductase and oxidase activity, the reaction mixture consisted of an MP solution (final concentration: 1 mM), an enzyme solution (mitochondrial and microsomal fractions), an NADPH generating system (final concentration: 1.3 mM NADP+, 3.3 mM G-6-P, 0.4 units/ml G-6-PDH, 3.3 mM MgCl2) and 100 mM Na-K phosphate buffer (pH 7.4).The enzyme solution was added to a one tenth volume of the reaction mixture. Various concentrations of GA (final concentrations: 0.005-1 μM) were dissolved in ethanol (final concentration: less than 2%), and were added to the reaction mixtures before preincubation for five min at 37 °C. After preincubation, an MP solution was added to the reaction mixture. The reactions were terminated by adding 2 volumes of methanol containing 3-benzoylpridine as an internal standard (IS) after 60 min and the mixtures were then cooled on ice for 30 min. After centrifugation (20,630 g for 5 min at 4 °C), the solutions were filtered using a Cosmonice filter S (pore size; 0.45 μm, filter diameter; 4 mm; Nacalai tesque, Tokyo, Japan) for HPLC analysis. Specific activities of the enzyme in the subcellular fractions were expressed as nanomoles of MP metabolites formed per min per mg of protein. Protein concentrations were determined by the Bicinchoninic acid methodwith bovine serum albumin as the standard [17].
Drug administration and sample preparation
The co-administration of GA to male rats was based on the method of Lin et al. [18]. The rats were divided into two groups and administered intraperitoneally with GA at a dose of 10 mg/kg or vehicle three times before MP dosing. The rats were fasted overnight before MP administration. After dissolving the MP in 5% glucose, the resulting MP solution to rats was administered at a single oral dose of 50 mg/kg. Whole blood samples were obtained from the abdominal aorta at 0.25, 0.5, 1, 2, 4, and 6 h after MP dosing and were immediately drawn into heparinized test tubes. Plasma samples were obtained by centrifugation (3,000 g for 10 min at 4 °C) of the blood samples and were stored at -80 °C until used in an analysis. The plasma (200 μL) was deproteinized by adding methanol containing IS (400 μL), and the mixtures allowed to stand for 30 min at room temperature. The HPLC samples were prepared by the same method as for the enzyme assay.
HPLC-ultraviolet (HPLC-UV) analysis
The HPLC-UV conditions were the same as used in our previous report [11]. Calibration curves were constructed by plotting the peakheight ratio of MP and its metabolites to that for IS in the range of 1.13 - 90.0 ng/mL(MPOL; 4.93-394 nmol/L, MP NOI and MP NOII; 4.64-372 nmol/L, MPOL NOI and MPOL NOII; 4.61 -368 nmol/L). The lower limit of quantification (LLOQ) was defined as the lowest concentration on the calibration curve. The data were collected and analyzed using GL Science software.
Pharmacokinetics analysis
The pharmacokinetic parameters of MP and its metabolites were estimated by a non-compartmental analysis were calculated using Phoenix WinNonlin (version 6.3, Pharsight, Mountain View, CA). The estimated parameters included the area under the plasma concentration-time curve from time 0 to 6 h (AUC6h), the area under the plasma concentration-time curve from time zero to infinite time (AUC∞) and the elimination half-life (t1/2) were determined. In addition, maximum plasma concentration (Cmax) and the time to reach the peak plasma concentration (tmax) were also determined.
Statistics
Data are presented as the mean value and standard deviation of the mean. In invitro MP metabolism, IC50 (concentration of inhibitor producing 50% inhibition in enzyme activity) for 11β-HSD1 inhibition by GA was calculated using nonlinear regression. In the pharmacokinetics of MP and its metabolites after MP alone and the co-administration dose of GA, statistical analysis between the groups was assessed by means of unpaired Student’s t test. A p-value of <0.05 was considered statistically significant.
Results
Inhibitory effects of GA on MP reduction activity in the liver
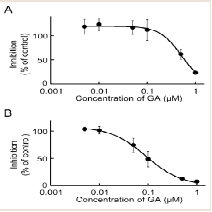
Our recent invitro study on MP metabolism in Wistar male rats suggested that MP was efficiently reduced to MPOL in the presence of an NADPH generation system in the liver, and that reduction of MP to MPOL was catalyzed by 11β-HSD1in microsomal and mitochondrial fractions [11]. The finding presented here in indicates that the NADPH-dependent MP reduction activities in the liver microsomal and mitochondrial fractions were 4.08 and 0.76 nmol/min/mg protein, respectively. When T0504 (1 μM), a selective 11β-HSD1 inhibitor, was added to the reaction system to identify the existence of 11β-HSD1 in both subcellular fractions, MPOL productions were inhibited by approximately 91% in both fractions (data not shown). Since T0504 is not drug but compound, we examined the issue of whether the reduction of MP in both subcellular fractions was inhibited by GA, a drug and a potent 11β-HSD1 inhibitor ((Figure 2A,2B)). When various concentrations of GA were added to the reaction systems, microsomal reduction of MP to MPOL was inhibited in a dose-dependent manner with an IC50value of 0.62 μM (Figure 2A). Similarly, GA inhibited mitochondrial MP reduction activity with the IC50value of 0.10 Μm (Figure 2B). These results suggest that reduction of MP to MPOL in both fractions in the liver were catalyzed by 11β-HSD1, and that the inhibitory strength was higher in the mitochondrial fraction than in the microsomal fraction.
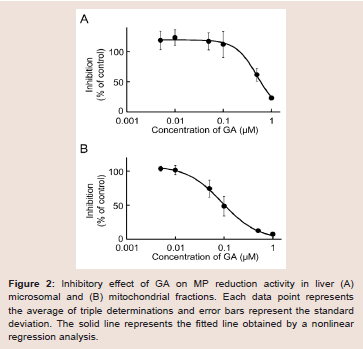
Figure 2: Inhibitory effect of GA on MP reduction activity in liver (A) microsomal and (B) mitochondrial fractions. Each data point represents the average of triple determinations and error bars represent the standard deviation. The solid line represents the fitted line obtained by a nonlinear regression analysis.
Inhibitory effects of GA on MP reduction activity in the testis
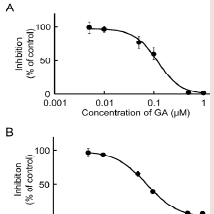
The rat testis was the secondary high tissue for the NADPHdependent MP reduction activities [11]. The activities in the cmicrosomal and mitochondrial fractions were 2.96 and 0.83 nmol/min/mg protein, respectively. Both activities were inhibited approximately 90% by T0504 (1 μM), suggesting that 11β-HSD1 was located in the testis (data not shown). The inhibitory effects of GA against the reduction of MP to MPOL in the testis were investigated in the same manner as the liver (Figure 3A,3B). As shown in (Figure 3), the testis microsomal and mitochondrial MP reductions were inhibited by addition of GA, and these inhibitions were dosedependent, with an IC50 value of 0.13 and 0.07 μM, respectively. These results indicate that MP reduction is also inhibited by GA in the testis.
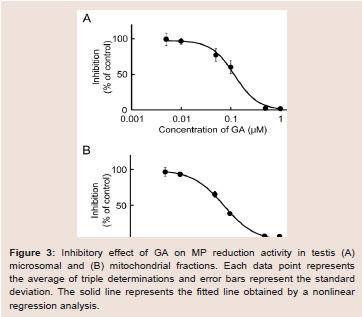
Figure 3: Inhibitory effect of GA on MP reduction activity in testis (A) microsomal and (B) mitochondrial fractions. Each data point represents the average of triple determinations and error bars represent the standard deviation. The solid line represents the fitted line obtained by a nonlinear regression analysis.
Interaction with GA on the pharmacokinetics of MP and MPOL
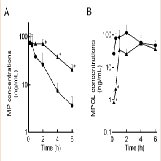
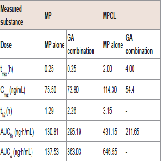
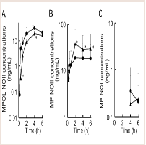
The effect of 11β-HSD1 inhibition by the co-administration of GA on the pharmacokinetics of MP and its metabolites in rats was investigated in an invivo study. (Figure 4 and Table 1) show the plasma concentration profiles and the pharmacokinetic parameters for MP and MPOL after the administration of MP to rats, with or without GA, respectively. Since it is known that GA rapidly inhibits 11β-HSD1 in invivo studies [18,19], the effect of the inhibition was evaluated for the periods of upto 6 h after the administration of MP. The co-administration of GA had no effect on the Cmax of MP at 0.25 h following an oral administration of MP, similar to MP alone, indicating that GA has no effect on the absorption of MP (Figure 4A and Table 1). After that, the t1/2of MP was approximately double as the result of the co-administration of GA, and a statistically significant increase was observed in the concentration profile from 2 h onward after the MP dose (Figure 4A). Thus, relative to the AUC of MP alone(130.81 ng∙h/mL), a 2.2-fold increase in that of co-administration of GA (295.19 ng∙h/mL) was observed (Table 1). On the other hand, the MPOL plasma concentration rapidly and significantly decreased as the result of the co-administration of GA compared with MP alone and then slowly increased within 4 h of tmax. The profile after tmax was the almost the same as that for MP alone (Figure 4B). As shown in (Table 1), the Cmax (54.4 ng/mL) and AUC6h (211.65 ng∙h/mL) after the co-administration of GA decreased to one half the level of those after MP alone (Cmax; 114.30 ng/mL and AUC6h; 431.15 ng∙h/mL).
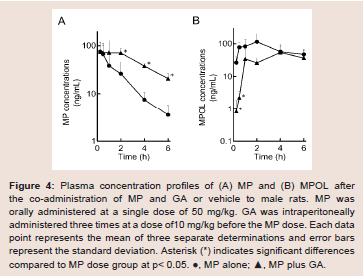
Figure 4: Plasma concentration profiles of (A) MP and (B) MPOL after the co-administration of MP and GA or vehicle to male rats. MP was orally administered at a single dose of 50 mg/kg. GA was intraperitoneally administered three times at a dose of10 mg/kg before the MP dose. Each data point represents the mean of three separate determinations and error bars represent the standard deviation. Asterisk (*) indicates significant differences compared to MP dose group at p< 0.05. ●, MP alone; ▲, MP plus GA.
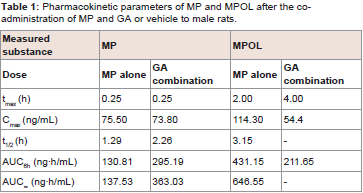
Table 1: Pharmacokinetic parameters of MP and MPOL after the coadministration of MP and GA or vehicle to male rats.
Interaction with GA on the pharmacokinetics n of MPOLNOII and MPNOII:
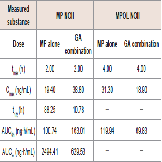
Since GA caused variations in the pharmacokinetics of MP and MPOL, we also examined the issue of whether the pharmacokinetics of other MP metabolites (MPOLNOI, MP NOI, MPOL NOII and MP NOII) were changed by the dose of GA. (Figure 5 and Table 2) depict the plasma concentration profiles and the pharmacokinetic parameters of MP NOI, MPOL NOII and MP NOII after the administration of MP with or without GA to the rats, respectively. The pharmacokinetics of MPOL NOI was not evaluated, because the plasma concentrations were LLOQ in both dose groups for all sampling points (data not shown). In the GA co-administration group, a significantly lower profile of MPOL NOII was immediately observed after the MP dose, similar to the MPOL profile, resulting in an LLOQ at 0.25 h (Figure 4B,5A). Due to the slow increase in the plasma concentration of MPOLNOII by the co-administration of GA, a significant difference was also observed at 4hafter the MP dose, and the Cmax and AUC6h (18.90 ng/mL and 69.83 ng∙h/mL) were around one half compared with the values for the MP alone group (31.30 ng/mL and 119.94 ng∙h/mL) (Figure 5A and Table 2). On the other hand, the co-administration with GA caused a statistically significant increase in the plasma concentration of MP NOII at 2 and 4h after the MP dose (Figure 5B). The Cmax and AUC6h (38.80ng/mL and 163.01 ng∙h/mL) were around twice and 1.6 times higher than those of MP alone group (19.40ng/mL and 100.74 ng∙h/mL), respectively (Table 2). On the other hand, GA had no observed effect on MP NOI plasma concentrations because of the low plasma concentration and large individual variability (Figure 5C).
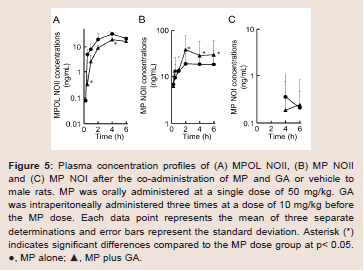
Figure 5: Plasma concentration profiles of (A) MPOL NOII, (B) MP NOII and (C) MP NOI after the co-administration of MP and GA or vehicle to male rats. MP was orally administered at a single dose of 50 mg/kg. GA was intraperitoneally administered three times at a dose of 10 mg/kg before the MP dose. Each data point represents the mean of three separate determinations and error bars represent the standard deviation. Asterisk (*) indicates significant differences compared to the MP dose group at p< 0.05. ●, MP alone; ▲, MP plus GA.
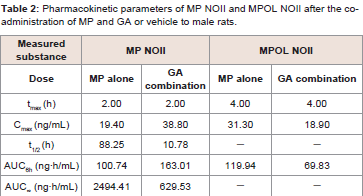
Table 2: Pharmacokinetic parameters of MP NOII and MPOL NOII after the coadministration of MP and GA or vehicle to male rats.
Discussion
In this study, we demonstrated that the reduction of MP to MPOL, a major MP metabolite, was inhibited by GA, an inhibitor of 11β-HSD1, in the microsomal and mitochondrial fractions of the liver and testis in Wistar male rats, and that the plasma pharmacokinetics of MP and its metabolites were modulated by the co-administration of GA.
Our previous report provided evidence that suggested MPOL was produced mainly in the liver, in which the major enzyme was 11β-HSD1, based on the inhibitor sensitivities [11]. However, the effect of an 11β-HSD1 inhibitor on MP reduction in the testis and the pharmacokinetics of MP remains unclear. Although it was reported that some drugs inhibit 11β-HSD1 [15,17], we focused on GA which has the strongest inhibitory effect of 11β-HSD1 with a minimum concentration as a possible drug in combination with MP. Since GA strongly inhibits NADPH-dependent 11β-HSD1 activity in the liver and testis [19], which MPOL was mainly produced, the drug was a good compound for evaluating drug-drug interactions towards MP metabolism. Furthermore, Wistar male rats were a relevant species for use in predicting drug-drug interactions related to 11β-HSD1 in humans, because the primary enzyme for reduction of MP to MPOL is 11β-HSD1 in the liver microsomal and mitochondrial fractions, similar to humans [11]. In the present study, the concentration of GA were determined based on the inhibition constant (Ki) for 11β-HSD1- dependent metabolism of cortisone to cortisol (active form) [15]. GA inhibited mitochondrial MP reduction activity (IC50:0.10 μM) stronger than microsomal activity (IC50; 0.62 μM) in the rat liver (Figure 2A,2B). Similarly, microsomal and mitochondrial MP reductions from the testis samples were inhibited by GA (IC50; 0.13 and 0.07 μM, respectively), suggesting that the testis enzyme involved in the reduction was also 11β-HSD1 ] ((Figure 3A,3B)). TheseIC50values were similar to Ki (0.44 μM)for GA against11β-HSD1 activity [15]. Although GA inhibits NADPH-dependent 11β-HSD1 and NAD+-dependent 11β-HSD2, which is highly expressed in the liver and kidney, respectively [1,5-7], taking into account the major tissues for its metabolism (liver and 4 testis) and the cofactor(NADPH but not NAD+; data not shown) involved in the reduction of MP to MPOL, these data suggest that GA inhibits the 11β-HSD1- dependent reduction in these subcellular fractions of the liver and testis. Therefore, we then investigated the issue of whether the plasma pharmacokinetics of MP and its metabolites were modulated by the co-administration of GA with MP in invivo rat study.
Lin et al. reported that the pharmacokinetics of cortisone and its active metabolite, cortisol, are modulated by three doses daily of GA [18]. We used the same dose and regimen to evaluate the effect of co-administration of GA on the pharmacokinetics of MP and its metabolites. Interestingly, the co-administration of GA caused a rapid and significant decrease in the plasma concentration of MPOL after MP dosing, resulting in the Cmax and AUC6h of MPOL after MP with a GA dose to be twice lower than the values in the case for MP alone(Figure 4B and Table 1). The decrease in the parameters suggests that 11β-HSD1 is inhibited by GA. On the other hand, the plasma MPOL levels after 4 h were almost the same levels as those for MP alone(Figure 4B). This observation is consistent the rapid disappearance of 11β-HSD1 inhibition effect by GA [18,19], and implies that the inhibitory effect against the MP reduction by the co-administration of GA had a relatively short life time invivo. In contrast to the MPOL profiles, the plasma concentration of MP was significantly increased at 2 h after the MP dose by the co-administration of GA, resulting in around a two times increase in the Cmax and AUC6hfor MP (Figure 4A and Table 1). In addition, the co-administration of GA extended the t1/2of MP in plasma (Table 1). These results suggest that the reduction and pharmacokinetics of MP are affected by coadministration of GA as with those of a glucocorticoid, and that 11β-HSD is involved in the reduction of MP in rats. Furthermore, the plasma pharmacokinetics of MPOLNOII and MPNOII were also modulated by the co-administration of GA. A significant decrease was immediately observed in the plasma levels of MPOLNOII after MP with a GA dosing, similar to that which was observed in MPOL, resulting in a lowering in the Cmax and AUC6hvalues around half of those after MP alone (Figure 5A and Table 2).On the other hand, a significant increase was observed in the plasma concentration of MP NOII from 2 to 4 h after MP with the GA dose, resulting increase in the Cmax and AUC6hof 2 and 1.6 times, respectively, compared with MP alone (Figure 5B and Table 2).Since MP NOII production was inhibited by a CYP inhibitor (ketoconazole and quercetin) [11], the CYP-dependent oxidation of MP would become dominant, due to the inhibition of 11β-HSD1 by GA. Contrary to the invivo results, the production of MP NOII in the microsomal and mitochondrial fractions of the liver and testis were not enhanced by the addition of GA to the reaction system in the invitro study (data not shown). Therefore, the increase in the plasma MP NOII concentration can be attributed to drug-drug interactions in the subcellular fractions of other tissues. Regardless of the significant increase in the MP NOII plasma concentration by MP as the result of the GA dose, MP plasma concentration was also significantly increased. The discrepancy indicates that the ratios of MPOL and MPOL NOII production inhibition were higher than that for the increase in MP NOII production. When the abundance ratio and sum of the plasma concentrations of MP and its metabolites were calculated at each sampling point in the absence and presence of GA dose, there were no significant differences in the sum of concentrations at any sample point, and the decrease in the ratios of MPOL and MPOL NOII concentrations were higher than the increase in the ratio of MP NOII by MP with the GA dose (Figure 4B,5A and 5B). These may be due to the fact that no apparent decrease was observed in the MP plasma concentration. In previous report, we proposed that MPOL NOII was produced via MPOL but not MP NOII, because MPOL was the major metabolite in plasma, but was excreted only small amounts in urine and MPOL NOII was the major metabolite in urine [11]. The changes in the plasma concentration profiles of MPOL and MPOL NOII in the case of co-administration of GA seemed to support this hypothesis. In addition, this proposed metabolic pathway was the same as that reported by Martini et al. [20]. To predict drug-drug interaction that might be effected in humans, the MP metabolites produced in invivo were estimated based on the proposed pathway. Since MPOL and MP NOII were produced in the liver subcellular fractions of humans [11], MPOL NOII would also be expected to be produced in humans invivo. Therefore, the co-administration of MP with an 11β-HSD1- inhibiting drug may affect the pharmacokinetics of MP and its three metabolites (MPOL, MPOL NOII and MP NOII) in humans. Regarding the pharmacological activity, although MPOL is the active metabolites with the same activity as MP in humans [21], the plasma concentration was significantly decreased by GA, contrary to the significant increase of MP plasma concentration. In addition, there is no information that MPOL NOII and MP NOII have the activity. Considering that the extensive difference was not observed in the sum of AUC6h of MP and MPOL between MP alone and GA combination, even though the pharmacokinetics of MP and its metabolites were modified by 11β-HSD1 inhibitor, suggesting a low potential to affect the pharmacological activity of MP. Recently, drugs which inhibit 11β-HSD1 have been developed to treat obesity [22-24]. From the present evidence, it would be expected that the administration of MP with an 11β-HSD1-inhibiting drug to humans may modulate the pharmacokinetics of MP and its metabolites. Consequently, our results represented the useful information for predicting the drugdrug interaction in humans.
Conclusions
The findings of this study demonstrate that GA inhibits the reduction of MP to MPOL at the low concentrations in the microsomal and mitochondrial fractions of, not only the liver but also the testis in Wistar male rats, suggesting that 11β-HSD1 contributes to MP reduction. Based on the variation in the plasma pharmacokinetics of MP and its metabolites by the co-administration of MP with GA in rats, the findings suggest that an 11β-HSD1 inhibitor, such as GA, could modulate the pharmacokinetics of this reduction pathway.
References
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, et al. (2004) 11β-hydroxysteroid dehydrogenase type 1 a tissue-specific regulator of glucocorticoid response. Endocr Rev 25: 831-866.
- Hoffmann F, Maser E (2007) carbonyl reductases and pluripotent hydroxysteroid dehydrogenases of the short-chain dehydrogenase/reductase superfamily. Drug Metab Rev 39: 87-144.
- Hult M, Jörnvall H, Oppermann UC (1998) Selective inhibition of human type 1 11beta-hydroxysteroid dehydrogenase by synthetic steroids and xenobiotics. FEBS Lett 441: 25-28.
- Mazzocchi G, Rossi GP, Neri G, Malendowicz LK, Albertin G, et al. (1998) 11beta-hydroxysteroid dehydrogenase expression and activity in the human adrenal cortex. FASEB J 12: 1533-1539.
- Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, et al. (1998) Immunohistochemical localization of type 1 11β-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab 83: 1325-1335.
- Maser E, Völker B, Friebertshäuser J (2002) 11 Beta-hydroxysteroid dehydrogenase type 1 from human liver: dimerization and enzyme cooperativity support its postulated role as glucocorticoid reductase. Biochemistry 41: 2459-2465.
- Seckl JR, Walker BR (2001) Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 142: 1371-1376.
- Kannisto K, Pietiläinen KH, Ehrenborg E, Rissanen A, Kaprio J, et al. (2004) Overexpression of 11beta-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J Clin Endocrinol Metab 89: 4414-4421.
- van Rossum EF, Lamberts SW (2006) Glucocorticoid resistance syndrome: a diagnostic and therapeutic approach. Best Pract Res Clin Endocrinol Metab 20: 611-626.
- Monaghan PJ, Owen LJ, Trainer PJ, Brabant G, Keevil BG, et al. (2011) Comparison of serum cortisol measurement by immunoassay and liquid chromatography-tandem mass spectrometry in patients receiving the 11β-hydroxylase inhibitor metyrapone. Ann Clin Biochem 48: 441-446.
- Murata H, Higuchi T, Otagiri M (2016) Oral pharmacokinetics and in-vitro metabolism of metyrapone in male rats. J Pharm Pharmacol 68: 970-979.
- Rosemond MJ, Walsh JS (2004) Human carbonyl reduction pathways and a strategy for their study in vitro. Drug Metab Rev 36: 335-361.
- Testa B, Jenner P (1981) Inhibitors of Cytochrome P-450s and their mechanism of action. Drug Metab Rev 12: 1-117.
- Shaw PN, Tseti J, Warburton S, Adedoyin A, Houston JB (1986) Inhibition of antipyrine metabolite formation. Steady state studies with cimetidine and metyrapone in rats. Drug Metab Dispos 14: 271-276.
- Arampatzis S Kadereit B, Schuster D, Balazs Z, Schweizer RA, et al. (2005) Comparative enzymology of 11beta-hydroxysteroid dehydrogenase type 1 from six species. J Mol Endocrinol 35: 89-101.
- Lin D, Sun W, Wang Z, Chen LG, Chen XL, et al.(2012) The effect of glycyrrhetinic acid on pharmacokinetics of cortisone and its metabolite cortisol in rats. J Biomed Biotechnol 2012: 856324.
- Diederrich S, Grossmann C, Hanke B, Quinkler M, Herrmann M, et al. (2000) In the search for specific inhibitors of human 11beta-hydroxysteroid-dehydrogenases (11beta-HSDs): chenodeoxycholic acid selectively inhibits 11beta-HSD-I. Eur J Endocrinol 142: 200-207.
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, et al. (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85.
- Marandici A, Monder C (1993) Inhibition by glycyrrhetinic acid of rat tissue 11beta-hydroxysteroid dehydrogenase in vivo. Steroids 58: 153-156.
- Martini R, Murray M (1996) Characterization of the invivo inhibition of rat hepatic microsomal aldehyde dehydrogenase activity by metyrapone. Biochem Pharmacol 51: 1187-1193.
- Meikle AW, West SC, Weed JA, Tyler FH (1975) Single dose metyrapone test: 11beta-hydroxylase inhibition by metyrapone and reduced metyrapone assayed by radioimmunoassay. J Clin Endocrinol Metab 40: 290-295.
- Byun SY, Shin YJ, Nam KY, Hong SP, Ahn SK (2015) A novel highly potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, UI-1499. Life Sci 120: 1-7.
- Hamilton BS, Himmelsbach F, Nar H, Schuler-Metz A, Krosky P, et al. (2015) Pharmacological characterization of the selective 11β-hydroxysteroid dehydrogenase 1 inhibitor, BI 135585, a clinical candidate for the treatment of type 2 diabetes. Eur J Pharmacol 746: 50-55.
- Chen XQ, Shao LD, Pal M, Shen Y, Cheng X, et al. (2015) Hupehenols A-E, selective 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors from Viburnum hupehense. J Nat Prod 78: 330-334.