
Journal of Clinical & Medical Case Reports
Download PDF
Case Report
*Address for Correspondence: Tetsuhiko Yasuno, MD PhD, Department of Nephrology and Rheumatology, Fukuoka University School of Medicine, 7-45-1 Nanakuma Johhnan-ku, Fukuoka 814-0180, Japan, Tel: +81-92-801-1011; Fax: +81-92-873-8008; E-mail: yasuno9584@fukuoka-u.ac.jp
Citation: Yasunaga T, Yasuno T, Imaizumi S, Nakashima H. The Use of Multiplex Ligation-Dependent Probe Amplification Technology for Genetic Analysis of the Entire PKD1 Gene and PKD2 Gene. J Clin Med Case Reports. 2015;2(1): 3.
Copyright © 2015 Yasuno et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 1
Submission: 10 February 2015 | Accepted: 22 April 2015 | Published: 27 April 2015


The Use of Multiplex Ligation- Dependent Probe Amplification Technology for Genetic Analysis of the Entire PKD1 Gene and PKD2 Gene
Tomoe Yasunaga1, Tetsuhiko Yasuno1*, Satoshi Imaizumi2 and Hitoshi Nakashima1
- 1Department of Nephrology and Rheumatology, Fukuoka University School of Medicine, Fukuoka, Japan
- 2Department of Cardiology, Fukuoka University School of Medicine Fukuoka, Japan
*Address for Correspondence: Tetsuhiko Yasuno, MD PhD, Department of Nephrology and Rheumatology, Fukuoka University School of Medicine, 7-45-1 Nanakuma Johhnan-ku, Fukuoka 814-0180, Japan, Tel: +81-92-801-1011; Fax: +81-92-873-8008; E-mail: yasuno9584@fukuoka-u.ac.jp
Citation: Yasunaga T, Yasuno T, Imaizumi S, Nakashima H. The Use of Multiplex Ligation-Dependent Probe Amplification Technology for Genetic Analysis of the Entire PKD1 Gene and PKD2 Gene. J Clin Med Case Reports. 2015;2(1): 3.
Copyright © 2015 Yasuno et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 1
Submission: 10 February 2015 | Accepted: 22 April 2015 | Published: 27 April 2015
Abstract
Genetic screening of the PKD1 and PKD2 genes using the MLPA method may decrease cost and labor. Indeed, the MLPA method was especially useful in cases exhibiting juvenile onset and rapid kidney enlargement. Case 1 had greater kidney capacity than the other cases, and dialysis was introduced at a younger age. There were two gene variations in PKD1, and the cyst in this case grew quite rapidly. Because the kidney volume was small in Case 2, the patient was not eligible for tolvaptan therapy according to guidelines in Japan. By using this method, it may be possible to predict ADPKD onset and the efficacy of tolvaptan, which is expected to suppress cyst growth, during the early stages of the disease.The use of multiplex ligation-dependent probe amplification technology for genetic analysis of the entire PKD1 gene and PKD2 gene.
Introduction
In common cases of autosomal dominant polycystic kidney disease (ADPKD), diagnosis is relatively simple using a combination of diagnostic imaging of the kidney and analysis of ADPKD in the patient’s family history. Therefore, in most patients with ADPKD, genetic testing for ADPKD is not necessary for diagnosis [1]. However, in ADPKD cases where a definitive diagnosis is not obtained by imaging or living relative related donor is identified for kidney transplantation, genetic testing may be necessary.The ADPKD is caused by mutations in one of two genes, PKD1 or PKD2. As for the variation of PKD1, cyst expansion and nephropathic progress are early than PKD2. Currently, genetic analysis is generally performed by direct sequencing of exons and proximal introns in genomic DNA using the Sanger method, and a number of genetic mutations in the PKD1 and PKD2 genes have been reported [2,3]. Sanger sequencing method may be effective in sporadic cases [4]. However, because these mutations are distributed throughout both genes, the entire sequence of each gene needs to be analyzed when a new family with ADPKD is screened, which is an expensive and time-consuming procedure. Although recent studies have used next-generation sequencing for such analyses, this method may be difficult in cases with high homology between the PKD1 gene and its pseudogenes.
Multiplex ligation-dependent probe amplification (MLPA) kits have been developed for genetic analysis by MRC-Holland using proprietary MLPA technology. A number of successful studies have reported using these kits, and they have been broadly applied to study hereditary tumors and other genetic diseases including large delitions, and to analyze tumor tissue and methylation levels [5]. Because MLPA kits (P351-B2, P352-C1) can efficiently analyze wide sequence ranges (e.g., from exons to chromosomes), they are suitable for genetic analysis of ADPKD, and some studies of genetic analysis of ADPKD using the MLPA method have been reported [6,7]. Thus, the MLPA method is thought to be useful when large genetic deletions are not detectable by the Sanger method.
Case Report
Despite the recent use of MLPA for the diagnosis of ADPKD, no studies have reported the genetic analysis of ADPKD in Japan population using the MLPA method. Therefore, in the present study, we analyzed the genetic characteristics of patients with ADPKD using MLPA. We obtained informed consent from nine patients (mean age, 45 years) and collected 8 mL of blood from each patient for analysis of genetic mutations in PKD1 and PKD2 genes using the MLPA method [5]. In case 1 (a 39-year-old man), we found deletions in exons 3 and 39 of the PKD1 gene (Figure 1). The kidney was extracted due to enlargement and cyst infection. Dialysis was initiated when the patient was 39 years of age. The kidney capacity was 5013 mL (Figure 2). A review of the family history revealed that the patient’s father had undergone maintenance hemodialysis and had died at the age of 46 years. In case 2 (a 32-year-old woman), we found a deletion in exon 3 of the PKD1 gene (Figure 1). Renal function was maintained, and the serum creatinine level was 0.7 mg/dL. She has hope of the delivery. Tolvaptan does not have the safety about the dosage to a pregnant woman. Tolvaptan was administered after delivery. The patient’s father had been diagnosed with ADPKD. No mutations were identified in the other subjects (mean age at which dialysis was initiated: 55.4±12.1 years; mean kidney capacity: 2592±1382mL).
Figure 1: Calculated probes ratios of the test sample. The probes indicated by arrow heads shows lower signal than other indicating reduced copy number in these particular chromosomal location.
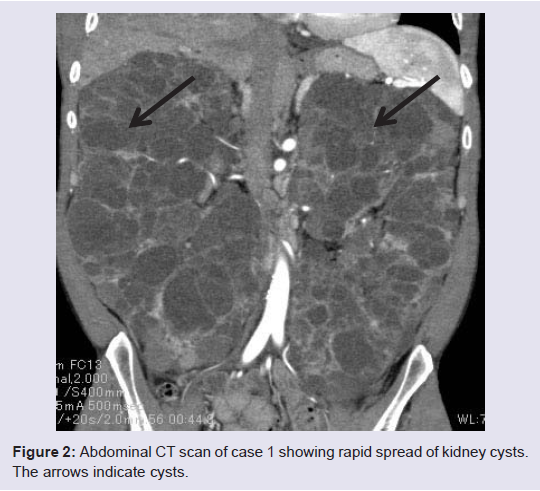
Figure 2: Abdominal CT scan of case 1 showing rapid spread of kidney cysts. The arrows indicate cysts.
Discussion
The juvenile onset and kidney rapid enlargement are symptoms of ADPKD in Case 1 and Case 2. In this analysis, both cases in which genetic deletions were detected exhibited juvenile onset and rapid kidney enlargement. In approximately 10% of ADPKD patients, mutations in the PKD1 or PKD2 genes cannot be identified byconventional genetic analysis of exons, such as the Sanger method. There are several potential reasons for this. First, some mutations are found in the introns or promoters distant from exons. Moreover, among patients diagnosed with ADPKD, mutations are generally found in genes responsible for diseases that cause multiple cysts in the kidney; for example, the HNF1βand PRKCSH genes, encoding hepatocyte nuclear factor 1β and protein kinase C substrate 80K-H, respectively, have been shown to be mutated [8]. Mutations have also been shown to reduce the function of the produced polycystin-1 protein, rather than eliminating the protein all together; such mutations included missense mutations in the PKD1 gene and cases where there is mosaicism of normal cells and cells with mutations in the PKD1 or PKD2 genes [9]. Thus, in such cases, genetic mutations may not be identified.Genetic screening of the PKD1 and PKD2 genes using the MLPA method may decrease cost and labor of routinely run diagnostics [5]. Indeed, the MLPA method was especially useful in cases exhibiting juvenile onset in twenties and rapid kidney enlargement that the extraction of the kidney is necessary. Case 1 had greater kidney capacity than the other cases, and dialysis was introduced at a younger age. There were two gene variations in PKD1, and the cyst in this case grew quite rapidly. Because the kidney volume was small in Case 2, the patient was not eligible for tolvaptan therapy according to guidelines in Japan.
There are some specific mutations tested in MLPA assays that predispose to ADPKD. The deletion of single exons detected by MLPA should be verified by other method (e.g. Sanger sequencing) to avoid false positive results due to the probe artifact, especially for exon 39 deletion reported only in one sample [7]. By using this method, it may be possible to predict ADPKD onset and the efficacy of tolvaptan that the treatment from an early stage is necessary, which is expected to suppress cyst growth, during the early stages of the disease.
References
- Gabow PA (1993) Autosomal dominant polycystic kidney disease. N Engl J Med 329: 332-342.
- Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, et al. (2007) Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 18: 2143-2160.
- Garcia-Gonzalez MA, Jones JG, Allen SK, Palatucci CM, Batish SD, et al. (2007) Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol Genet Metab 92: 160-167.
- Audrézet MP, Cornec-Le Gall E, Chen JM, Redon S, Quéré I, et al. (2012) Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat 33: 1239-1250.
- Stuppia L, Antonucci I, Palka G, Gatta V (2012) Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int J Mol Sci 13: 3245-3276.
- Consugar MB, Wong WC, Lundquist PA, Rossetti S, Kubly VJ, et al. (2008) Characterization of large rearrangements inautosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 74: 1468-1479.
- Obeidova L, Elisakova V, Stekrova J, Reiterova J, Merta M, et al. (2014) Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease. BMC Med Genet 15: 41.
- Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, et al. (2011) Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 22: 2047-2056.
- Consugar MB, Wong WC, Lundquist PA, Rossetti S, Kubly VJ, et al. (2008) Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 74: 1468-1479.