
Journal of Vaccine & Immunotechnology
Download PDF
Research Article
*Address for Correspondence: Akira Matsumoto, Department of Infection and Host Defenses, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan, Tel: +81-89-960-5274; Fax: +81-89-960-5275; E-mail: akiramat@m.ehime-u.ac.jp
Citation: Matsumoto A, Maruyama S, Takahashi S, Miyoshi Y, Kanoh M, et al. Autophagic Control of Listeria Determines the Infection-Induced Death of Macrophages. J Vaccine Immunotechnology. 2013;1(1): 6.
Copyright © 2013 Matsumoto A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 15 June 2013 | Accepted: 17 July 2013 | Published: 19 July 2013
Autophagic Control of Listeria Determines the Infection-Induced Death of Macrophages
Akira Matsumoto1*, Saho Maruyama2, Saori Takahashi1, Yukiyo Miyoshi1, Makoto Kanoh1, Makoto Kuwahara2,3, Takeshi Yamada1, Masakatsu Yamashita2 and Yoshihiro Asano1,2,4
- 1Department of Infection and Host Defenses, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
- 2Department of Immunology, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
- 3Translational Research Center, Ehime University Hospital, Toon, Ehime, Japan
- 4Laboratory for Cytokine Regulation, RIKEN Integrated Medical Science Center, Tsurumi, Yokohama, Kanagawa, Japan
*Address for Correspondence: Akira Matsumoto, Department of Infection and Host Defenses, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan, Tel: +81-89-960-5274; Fax: +81-89-960-5275; E-mail: akiramat@m.ehime-u.ac.jp
Citation: Matsumoto A, Maruyama S, Takahashi S, Miyoshi Y, Kanoh M, et al. Autophagic Control of Listeria Determines the Infection-Induced Death of Macrophages. J Vaccine Immunotechnology. 2013;1(1): 6.
Copyright © 2013 Matsumoto A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 15 June 2013 | Accepted: 17 July 2013 | Published: 19 July 2013
Abstract
Listeria induces cell death in macrophages, hepatocytes and dendritic cells. It is accepted that the cell death is induced by listeriolysin O (LLO), which has a crucial role in the escape of Listeria from the vacuole. To re-evaluate the requirement of LLO for inducing cell death, we used an hlyW492A mutant that possesses a mutation in codon Trp-492 of LLO, with leads to a 1000-fold decrease in the hemolytic activity. Although the hlyW492A mutant possesses decreased hemolytic activity, it can still escape from the phagosome into the cytosol and induces cell death in macrophages. An aroA-deficient mutant that has normal LLO activity and a reduced replication capacity in the cytosol because of the deficiency of menaquinone failed to induce cell death in macrophages. The addition of menaquinone, which is a component of the electron-transfer pathway, accelerated the cytotoxicity and bacterial growth in aroA-deficient mutant-infected macrophages. These results indicate that the Listeria-infected cell death in macrophages depends on the cytosolic Listeria numberin addition to the LLO activity. Although the autophagy system fails to exclude wild-type Listeria from the infected macrophages, the aroA-deficient mutant was co-localized with autophagy marker LC3, suggesting the involvement of an autophagic mechanism to rescue the host cells when Listeria grows slowly in the cytosol. In cases with the growth of wild-type Listeria, the intermediate substances of oxidative metabolism or the rapid replication of Listeria might allow for an escape from the autophagy machinery, which induces death in the host cells. Therefore, our findings suggest that LLO is necessary for the bacteria to invade the cytosol, but is not sufficient for inducing Listeria-infected cell death in macrophages. An autophagy system apparently rescues host cells from infection-induced cell death even in the presence of LLO when the Listeria is growing slowly in the cytosol.Keywords
Listeria; Infection-induced cell death; hly; aroA; AutophagAbbreviations
BMMs: Bone Marrow Derived Macrophages; CFU: Colony-Forming Units ; LLO: Listeriolysin O; TGC-PEC: Thioglycollate-Elicited Peritoneal MacrophagesIntroduction
The Gram-positive intracellular bacterium Listeria monocytogenes (Lm) has been used as a classic model to study immune responses against intracellular bacteria [1-3]. Host cell infection begins with the internalization of the bacteria either by phagocytosis in the case of macrophages, or by induced phagocytosis in the case of nonphagocytic cells [4]. Following internalization, Lm resides within the phagosome before lysing the membrane. Within a half hour, Lm lyses the vacuolar membrane. This event is achieved by a potent poreforming toxin, LLO [5,6]. Mutant bacteria not expressing LLO are avirulent in the mouse, demonstrating that the escape of the bacteria to the cytosol is critical for the establishment of an infection [5]. After escape into the cytosol, the bacteria replicate and concomitantly become covered with actin filaments. When moving bacteria come in contact with the plasma membrane, they induce the formation of bacterium-containing protrusions. Contact between these protrusions and neighboring cells results in the internalization of the protrusion [6]. Therefore, once Lm has entered the cytoplasm, it can disseminate directly from cell to cell.Apart from its crucial role in the escape from the vacuole, LLO is also able to induce apoptosis [7]. It has been reported that Lm induces cell death in macrophages, hepatocytes and dendritic cells, and that this phenomenon involves LLO [4,5,8]. The type of cell death induced by Lm involves a variety of complex mechanisms, including apoptosis, necrosis or pyroptosis depending on cell type [9,10].
Pathogen-infected host cells utilizean autophagy system as a defense against intracellular pathogens [11-13]. Autophagy is a ubiquitous degradation system in eukaryotic cells and contributes to the removal of damaged organelles and the degradation of misfolded protein aggregates [14]. Cytosolic Lm interacts with the host autophagy system and is surrounded by double-membrane vacuoles that are colocalized with microtubule-associated protein light chain 3 (LC3) [15-17]. ActA and the phospholipases (PlcA and PlcB) play important roles in the escape from the autophagy system [16-18].
During a study comparing intracellular Lm growth and the induction of cell death in macrophages, we noticed the presence of cell death mechanism in addition to the LLO in Lm-infected macrophages. In the present report, we demonstrate that LLO is necessary for Lm to invade into the cytosol, but is not sufficient for inducing Lm-infected cell death in macrophages. A mutant possessing decreased LLO activity induced cell death, while a mutant possessing normal LLO activity but a low replication rate in the cytosol failed to induce cell death in macrophages. The latter mutant appeared to be surrounded by an autophagosome, suggesting the involvement of an autophagic mechanism in rescuing the host cells from Lm-induced cell death when Lm grows slowly in the cytosol.
Materials and Methods
Mice and cellsBALB/c mice were purchased from the Clea Japan (Tokyo, Japan) and GFP-LC3 transgenic mice were provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan [19]. Animals were used in accordance with our institutional guidelines for animal experimentation. Thioglycollate-elicited peritoneal macrophages (TGC-PEC) were collected from mice by peritoneal lavage four days after intraperitoneal injection of 2 ml of 4% thioglycollate (Difco Laboratories, Detroit, MI). Bone marrowderived macrophages (BMMs) were prepared by cultivating bone marrow cells with 10% L929 cell culture supernatant. TGC-PEC, BMMs and mouse macrophage cell lines RAW264.7 and J774A.1 were cultured in RPMI 1640 medium supplemented with 2mM L-glutamine, 1mM sodium pyruvate, 1x nonessential amino acids, 50 μM 2-mercaptoethanol and 10% heat-inactivated FCS.
Bacteria and reagents
The wild-type (WT) Lm strain (EGDe strain) and hly mutants of Lm were provided by Dr. P. Cossart, Pasteur Institute, Paris, France. The mutants used in this study were the following: hly null mutant (Δhly), which is deficient in hemolytic activity; hlyW492A mutant (W492A), which has a point mutation in LLO from Trp- 492 to Ala and exhibits a 1000-fold reduced hemolytic activity and comparable growth rate as WT Lm [20]; aroA null mutant (ΔaroA), which is deficient in of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase and has a deficiency in the common amino acid biosynthesis pathway [21]. The mutant has a comparable level of hly gene expression and a 2-fold higher LLO activity in comparison to WT Lm [22]. Lm was cultivated overnight at 37° C in 2 ml brain heart infusion (BHI) broth (Difco Laboratories). The bacterial culture was diluted 1:20 in fresh BHI medium and grown to the mid-exponential phase. Menaquinone (Sigma-Aldrich, St. Louis, MO) was dissolved in isopropanol and added to the culture medium to a final concentration of 50 μg/ml. Shikimic acid (WAKO, Japan) and folic acid (Sigma- Aldrich) were dissolved in water and used at concentrations of 0.3 mg/ml and 0.1 mg/ml, respectively. Wortmannin (Sigma-Aldrich) was dissolved in dimethyl sulfoxide and diluted in culture media to achieve a final concentration of 100 nM.
Macrophage cytotoxicity assay
Cytotoxicity was determined by measuring the relative release of the cytosolic enzyme lactate dehydrogenase (LDH) from macrophages. At various time points, the LDH activity in macrophages was measured after lysis with 1% Triton X-100 using a kit purchased from Roche (Mannheim, Germany) according to the manufacturer’s protocol. Absorbance values at 490 nm were determined photometrically with a 96-well plate reader.
Bacterial infection and intracellular infection assays
TGC-PEC cells were seeded at 2 x 105 cells in 96-well plates containing 10% FCS-RPMI 1640 without antibiotics. After 3 h of cultivation, non-adherent cells were removed by washing with PBS. J774A.1 cells and RAW264.7 cells were seeded at 5 x 104 cells in 96-well plates and cultured for 24 h. Bacteria grown to the midexponential phase were washed, resuspended in cell culture medium and added to the cell monolayer at a MOI of 2-10. At this time point, the Lm infection was defined as t = 0. After 1 h incubation, the wells were washed and overlaid with fresh cell culture medium containing 50 μg/ml of Gentamicin (Sigma-Aldrich) to kill the extracellular bacteria. After the indicated periods, the cultures were washed again, lysed and homogenized with 0.05% Triton X-100 in PBS, and the number of colony-forming units (CFUs) of intracellular bacteria was determined.
Immunofluorescence microscopy
For the immunofluorescence analysis, cells were fixed by immersion in 4% paraformaldehyde in PBS. After 15 min, the fixed cells were further incubated with 1% FCS-PBS containing 0.2% Triton X-100 for 20 min. Cells were stained with DAPI (Dojindo, Japan) for 1 h. The stained samples were examined with a fluorescence microscope (Olympus, Japan).
Results
aroA-deficient Lm are less cytotoxic to macrophagesIt has been reported that Lm induces death in infected macrophages and dendritic cells [4,5,8]. The activity of the hly gene product, LLO, was shown to be essential for Lm-induced cell death [7]. To examine whether genes other than hly can induce cell death, we used the hly mutant Lm, hlyW492A, which possesses a mutation in codon Trp-492 of LLO, which is necessary for its hemolytic activity. The W492A mutation decreased the hemolytic activity in vitro by 99.9%, and the W492A mutant was reported to be 1000-fold less virulent in mice based on the LD50 [20]. After a 24 h Lm infection, the cytotoxicity of the bacteria to macrophages was determined by measuring the cytosolic LDH activityin the viable adherent RAW264.7cells. WT Lm and W492A mutant infections both induced cell death in RAW264.7 cells, while infection with the Δhly mutant failed to induce cell death (Figure 1 left). The number of CFU in the cytosol of cells infected with W492A mutant was equivalent to that of cells infected with WT Lm (data not shown). Although it has been reported that there is a correlation between Lm infection-induced cell death and the activity of LLO [20], the above results suggested the importance of the cytosolic invasion of Lm for the cytotoxicity.
 Figure 1: The W492A mutant, but not the ΔaroA mutant induces cytotoxicity in infected macrophages. The indicated macrophage cells and cell lines were infected with WT, W492A, Δhly and ΔaroA mutant, and the cytotoxicity of the Lm strain was determined by measuring the LDH activity as described in the Materials and Methods section.
Figure 1: The W492A mutant, but not the ΔaroA mutant induces cytotoxicity in infected macrophages. The indicated macrophage cells and cell lines were infected with WT, W492A, Δhly and ΔaroA mutant, and the cytotoxicity of the Lm strain was determined by measuring the LDH activity as described in the Materials and Methods section.
We screened many Lm mutants to further evaluate the genes involved in Lm infection-induced cell death. Among the Lm mutants screened, we found that the ΔaroA mutant failed to facilitate cell death in spite of their normal LLO activity and capacity to invade the cytosol invading capability in RAW267.4 and J774A.1 cells (Figure 1 left and middle) [21,22]. Infection of J774A.1 cells with WT Lm resulted in almost complete destruction of the cells (Figure 1 middle). We further evaluated the effect of the ΔaroA mutant on TGCinduced heterogeneous macrophage cells. As shown in Figure 1 right, the effect of ΔaroA mutant on TGC-PEC was similar to that on the RAW264.7 and J774A.1 cells. These results suggested that the ΔaroA mutant provides a good tool to examine what mechanisms other than LLO are involved in Lm infection-induced cell death.
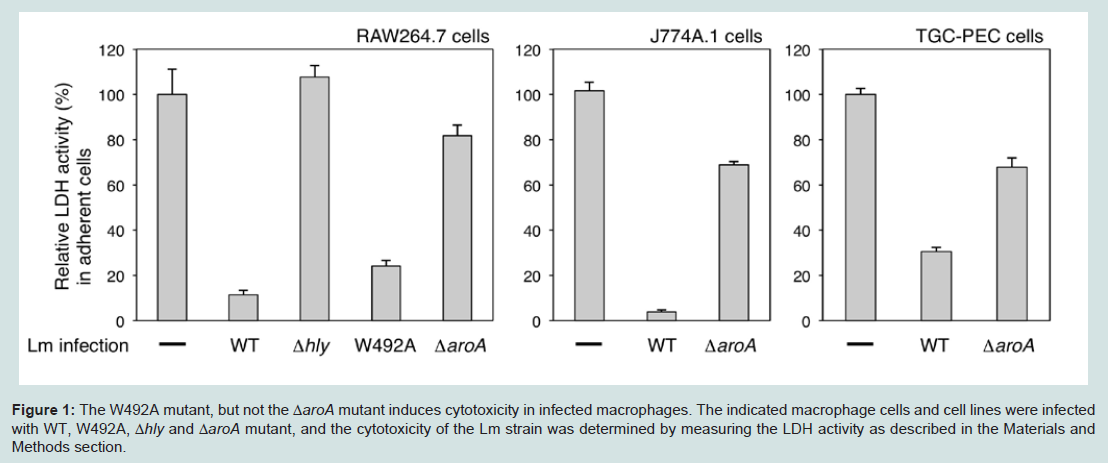 Figure 1: The W492A mutant, but not the ΔaroA mutant induces cytotoxicity in infected macrophages. The indicated macrophage cells and cell lines were infected with WT, W492A, Δhly and ΔaroA mutant, and the cytotoxicity of the Lm strain was determined by measuring the LDH activity as described in the Materials and Methods section.
Figure 1: The W492A mutant, but not the ΔaroA mutant induces cytotoxicity in infected macrophages. The indicated macrophage cells and cell lines were infected with WT, W492A, Δhly and ΔaroA mutant, and the cytotoxicity of the Lm strain was determined by measuring the LDH activity as described in the Materials and Methods section.Menaquinone accelerates the death of ΔaroA mutant infected macrophages
The aroA gene product is the first enzyme in the aromatic amino acid biosynthesis pathway in Lm (Figure 2A). It has been reported that the ΔaroA mutant exhibited an attenuated virulence in mice because of its slow replication and cell-to-cell spread. The ΔaroA mutant is unable to grow without aromatic acids in minimal medium. The addition of menaquinone (an essential component of the respiratory chain in gram-positive bacteria) to the minimal medium restores the growth of the ΔaroA mutant [21]. Since the ΔaroA mutant failed to induce cell death following infection, we evaluated whether the intermediate metabolites of the aroA-encoded enzyme could restore the outcome of the infection.
 Figure 2: Shikimic acid and menaquinone restore the cytotoxicity of the &Delat;aroA mutant. (A) aroA encodes 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase, which functions in the early phase of the common amino acid biosynthesis pathway in Lm. Adapted from Stritzker et al. [21] with modifications. Dashed arrows show several enzymes. (B & C) RAW264.7 cells were infected with WT, ΔaroA mutant or Δhly mutantin the presence or absence of the indicated reagents (M, menaquinone; S, shikimic acid; F, folic acid) and the cytotoxicity was determined. (D) TGC-PEC and J774A.1 cells were infected with WT or ΔaroA mutant in the presence or absence of either shikimic acid or menaquinone. The cytotoxicity was determined as described in the Materials and Methods section.
Figure 2: Shikimic acid and menaquinone restore the cytotoxicity of the &Delat;aroA mutant. (A) aroA encodes 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase, which functions in the early phase of the common amino acid biosynthesis pathway in Lm. Adapted from Stritzker et al. [21] with modifications. Dashed arrows show several enzymes. (B & C) RAW264.7 cells were infected with WT, ΔaroA mutant or Δhly mutantin the presence or absence of the indicated reagents (M, menaquinone; S, shikimic acid; F, folic acid) and the cytotoxicity was determined. (D) TGC-PEC and J774A.1 cells were infected with WT or ΔaroA mutant in the presence or absence of either shikimic acid or menaquinone. The cytotoxicity was determined as described in the Materials and Methods section.
We used an intermediate metabolite shikimic acid, and end products menaquinone and folic acid. These three compounds did not show any cytotoxicity when added to Δhly mutant-infected RAW264.7 cells (Figure 2B). However, there was dose-dependent effect of menaquinone in ΔaroA mutant-infected macrophages (Figure 2C). The addition of either shikimic acid or menaquinone to the culture induced cell death in ΔaroA mutant-infected macrophages (Figure 2D). These results suggested that the products of the aromatic amino acid biosynthesis pathway mediated by the aroA-encoded enzyme are important for inducing Lm infection-induced cell death in macrophages. Since the ΔaroA mutant is deficient in oxidative respiration due to the lack of menaquinone [21], this suggesting that the growth of Lm in the cytosol is essential for the Lm infectioninduced cell death.
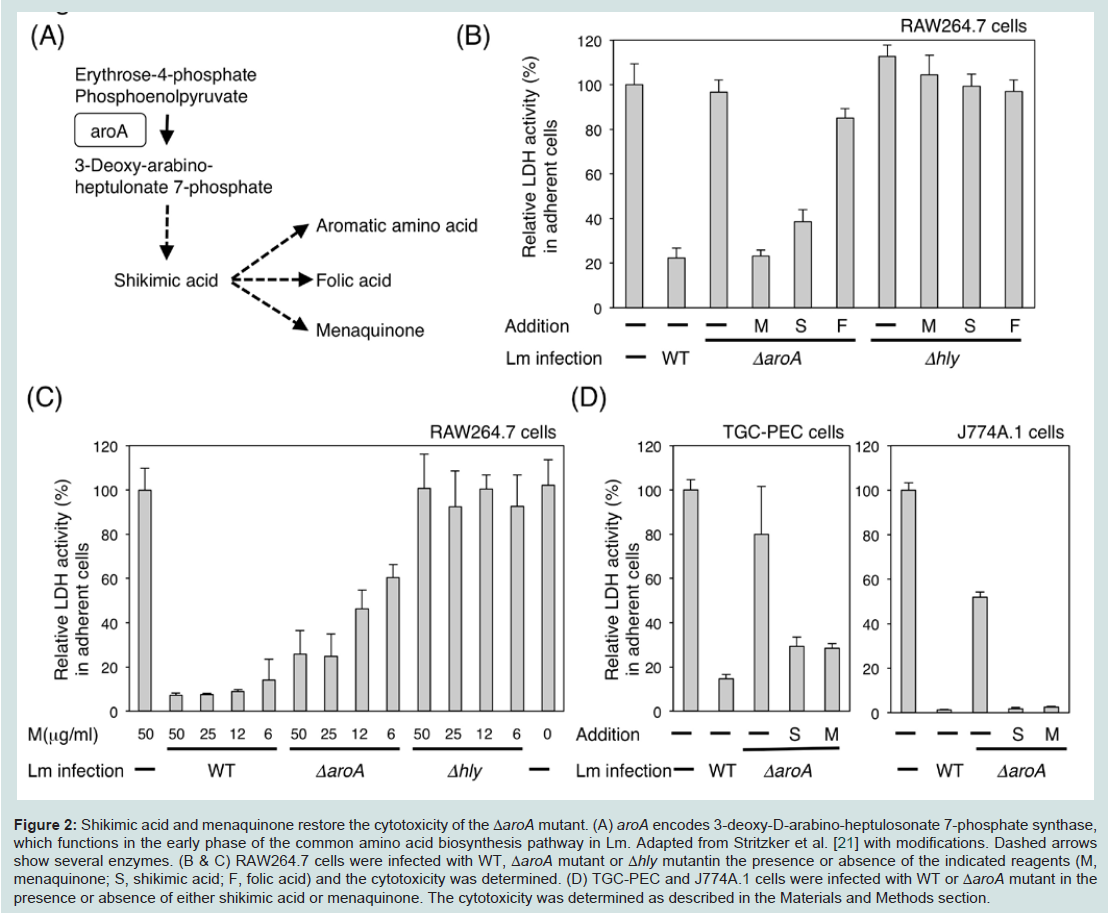 Figure 2: Shikimic acid and menaquinone restore the cytotoxicity of the &Delat;aroA mutant. (A) aroA encodes 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase, which functions in the early phase of the common amino acid biosynthesis pathway in Lm. Adapted from Stritzker et al. [21] with modifications. Dashed arrows show several enzymes. (B & C) RAW264.7 cells were infected with WT, ΔaroA mutant or Δhly mutantin the presence or absence of the indicated reagents (M, menaquinone; S, shikimic acid; F, folic acid) and the cytotoxicity was determined. (D) TGC-PEC and J774A.1 cells were infected with WT or ΔaroA mutant in the presence or absence of either shikimic acid or menaquinone. The cytotoxicity was determined as described in the Materials and Methods section.
Figure 2: Shikimic acid and menaquinone restore the cytotoxicity of the &Delat;aroA mutant. (A) aroA encodes 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase, which functions in the early phase of the common amino acid biosynthesis pathway in Lm. Adapted from Stritzker et al. [21] with modifications. Dashed arrows show several enzymes. (B & C) RAW264.7 cells were infected with WT, ΔaroA mutant or Δhly mutantin the presence or absence of the indicated reagents (M, menaquinone; S, shikimic acid; F, folic acid) and the cytotoxicity was determined. (D) TGC-PEC and J774A.1 cells were infected with WT or ΔaroA mutant in the presence or absence of either shikimic acid or menaquinone. The cytotoxicity was determined as described in the Materials and Methods section.Growth of the ΔaroA mutant in macrophages
To determine the correlation between cytosolic replication and Lm infection-induced cell death, we next examined the Lm survival rate in macrophages. The invasion of the W492A mutant and ΔaroA mutant from the phagosome into the cytosol was similar to that of the WT Lm. The growth of the W492A mutant was equivalent to that of the WT Lm, while the growth of the ΔaroA mutant was strongly reduced. The Δhly mutant was rapidly destroyed in BMMs (Figure 3A). The growth rate of the Lm in macrophages correlates with their cytotoxicity, as shown in Figure 1 left. In order to evaluate the influence of the aroA-deficiency in Lm-infected macrophages, we compared the growth rates of the different Lm strains in the cytosol of macrophages. The growth rate of the ΔaroA mutant in macrophages was slower than that of the other Lm (Figure 3A). However, the addition of shikimic acid promptly restored the replication of the ΔaroA mutant in the cytosol to an equivalent level as was seen for the WT Lm, while the addition of menaquinone restored the replication of ΔaroA mutant with some delay (Figures 3B-3D). In contrast, the addition of folic acid did not change the growth rate of the ΔaroA mutant in J774A.1 cells (Figure 3D). These results indicated that menaquinone is essential for the intracellular growth of Lm, and that the replication of Lm in the cytosol is an important factor for inducing the infection-mediated death of macrophages.
 Figure 3: The ΔaroA mutant showed a reduced growth rate in macrophages.(A) BMMs were infected with WT (open circle), Δhly mutant (open square), W492A mutant (closed diamond) and ΔaroA mutant (open triangle), and the CFUs at the indicated time points were calculated as described in the Materials and Methods section. (B & C) RAW264.7 cells and TGC-PEC cells were infected with WT (open circle) or ΔaroA mutant (open triangle) in the presence of additional shikimic acid (closed triangle) or menaquinone (closed square), and the CFUs at the indicated time points were calculated. (D) J774A.1 cells were infected with WT (open circle) in the presence of shikimic acid (asterisk), or ΔaroA mutant (open triangle) in the presence of shikimic acid (closed triangle), menaquinone (closed square) or folic acid (closed diamond), and the CFUs at the indicated time points were calculated.
Figure 3: The ΔaroA mutant showed a reduced growth rate in macrophages.(A) BMMs were infected with WT (open circle), Δhly mutant (open square), W492A mutant (closed diamond) and ΔaroA mutant (open triangle), and the CFUs at the indicated time points were calculated as described in the Materials and Methods section. (B & C) RAW264.7 cells and TGC-PEC cells were infected with WT (open circle) or ΔaroA mutant (open triangle) in the presence of additional shikimic acid (closed triangle) or menaquinone (closed square), and the CFUs at the indicated time points were calculated. (D) J774A.1 cells were infected with WT (open circle) in the presence of shikimic acid (asterisk), or ΔaroA mutant (open triangle) in the presence of shikimic acid (closed triangle), menaquinone (closed square) or folic acid (closed diamond), and the CFUs at the indicated time points were calculated.
High-replicating Listeria in the cytosol escapes from the autophagic control of intracellular pathogens
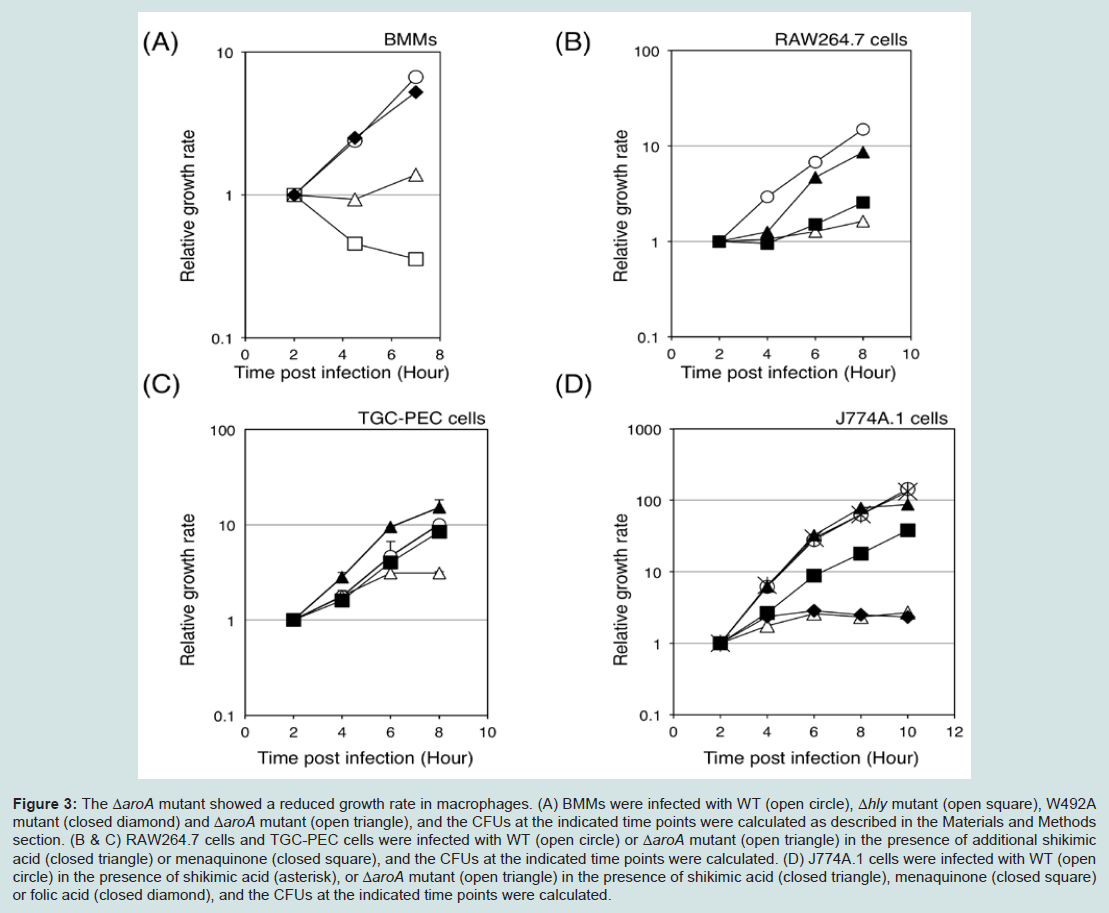 Figure 3: The ΔaroA mutant showed a reduced growth rate in macrophages.(A) BMMs were infected with WT (open circle), Δhly mutant (open square), W492A mutant (closed diamond) and ΔaroA mutant (open triangle), and the CFUs at the indicated time points were calculated as described in the Materials and Methods section. (B & C) RAW264.7 cells and TGC-PEC cells were infected with WT (open circle) or ΔaroA mutant (open triangle) in the presence of additional shikimic acid (closed triangle) or menaquinone (closed square), and the CFUs at the indicated time points were calculated. (D) J774A.1 cells were infected with WT (open circle) in the presence of shikimic acid (asterisk), or ΔaroA mutant (open triangle) in the presence of shikimic acid (closed triangle), menaquinone (closed square) or folic acid (closed diamond), and the CFUs at the indicated time points were calculated.
Figure 3: The ΔaroA mutant showed a reduced growth rate in macrophages.(A) BMMs were infected with WT (open circle), Δhly mutant (open square), W492A mutant (closed diamond) and ΔaroA mutant (open triangle), and the CFUs at the indicated time points were calculated as described in the Materials and Methods section. (B & C) RAW264.7 cells and TGC-PEC cells were infected with WT (open circle) or ΔaroA mutant (open triangle) in the presence of additional shikimic acid (closed triangle) or menaquinone (closed square), and the CFUs at the indicated time points were calculated. (D) J774A.1 cells were infected with WT (open circle) in the presence of shikimic acid (asterisk), or ΔaroA mutant (open triangle) in the presence of shikimic acid (closed triangle), menaquinone (closed square) or folic acid (closed diamond), and the CFUs at the indicated time points were calculated.Autophagy is an important host mechanism involved in the removal of intracellular bacteria and protozoan, which is in keeping with its primary function as a cytoplasmic clean-up process [16,23-25]. The above results suggested that an autophagic mechanism is involved in clearing the intracytoplasmic bacteria and rescues the host cells from infection-induced cell death. To directly evaluate the involvement of autophagy in preventing the cell death of Lm-infected macrophages, we utilized GFP-LC3 transgenic mice that express the GFP protein on LC3 molecules of the autophagosome [19]. DAPIpositive spots in ΔaroA mutant-infected TGC-PEC of GFP-LC3 transgenic mouse were co-localized with the GFP-positive spots, suggesting that the ΔaroA mutants were trapped by autophagosomes (Figure 4A). After the addition of menaquinone into the culture medium, DAPI-positive spots in the ΔaroA mutants were found to be free from GFP-LC3, thus suggesting that the ΔaroA mutants escaped from the autophagosomes when menaquinone was present (Figure 4A and 4B). To further evaluate the role of autophagic control, we monitored the cytosolic growth of ΔaroA mutants in the presence or absence of Wortmannin and menaquinone (Figure 4C). The result demonstrated that the growth of ΔaroA mutant was augmented in the presence of the autophagic inhibitor, Wortmannin. The addition of menaquinone strongly augmented the growth of the ΔaroA mutant, while the augmented growth of the ΔaroA mutant was not affected by the presence of the autophagic inhibitor. These results demonstrated that the autophagic system controls the cytosolic growth of the ΔaroA mutants. The addition of menaquinone and Wortmannin did not affect the growth of WT Lm (data not shown). These results suggest that highly-replicating Lm in the cytosol can escapes from the autophagic control of intracellular pathogens.
 Figure 4: The autophagy system controlled the ΔaroA mutant replication in the cytosol. (A) TGC-PEC cells of a GFP-LC3 transgenic mouse were infected for 1 h with the ΔaroA mutant and the cells were cultured for an additional 11 h in the presence or absence of menaquinone. After 12 h of Lm infection, the cells were fixed and stained with DAPI (blue), and were observed under the fluorescence microscopy. The DAPI spot co-localized with the GFP-LC3 spot is indicated with an arrowhead. (B) In order to quantify the co-localization of Lm and GFP-LC3 in (A), (C) J774A.1 cells were infected with ΔaroA mutant for 1 h. Menaquinone and Wortmannin (WTM) was added at 1 h post infection, and then the number of cytosolic bacteria was calculated at 15 h post infection.
Figure 4: The autophagy system controlled the ΔaroA mutant replication in the cytosol. (A) TGC-PEC cells of a GFP-LC3 transgenic mouse were infected for 1 h with the ΔaroA mutant and the cells were cultured for an additional 11 h in the presence or absence of menaquinone. After 12 h of Lm infection, the cells were fixed and stained with DAPI (blue), and were observed under the fluorescence microscopy. The DAPI spot co-localized with the GFP-LC3 spot is indicated with an arrowhead. (B) In order to quantify the co-localization of Lm and GFP-LC3 in (A), (C) J774A.1 cells were infected with ΔaroA mutant for 1 h. Menaquinone and Wortmannin (WTM) was added at 1 h post infection, and then the number of cytosolic bacteria was calculated at 15 h post infection.
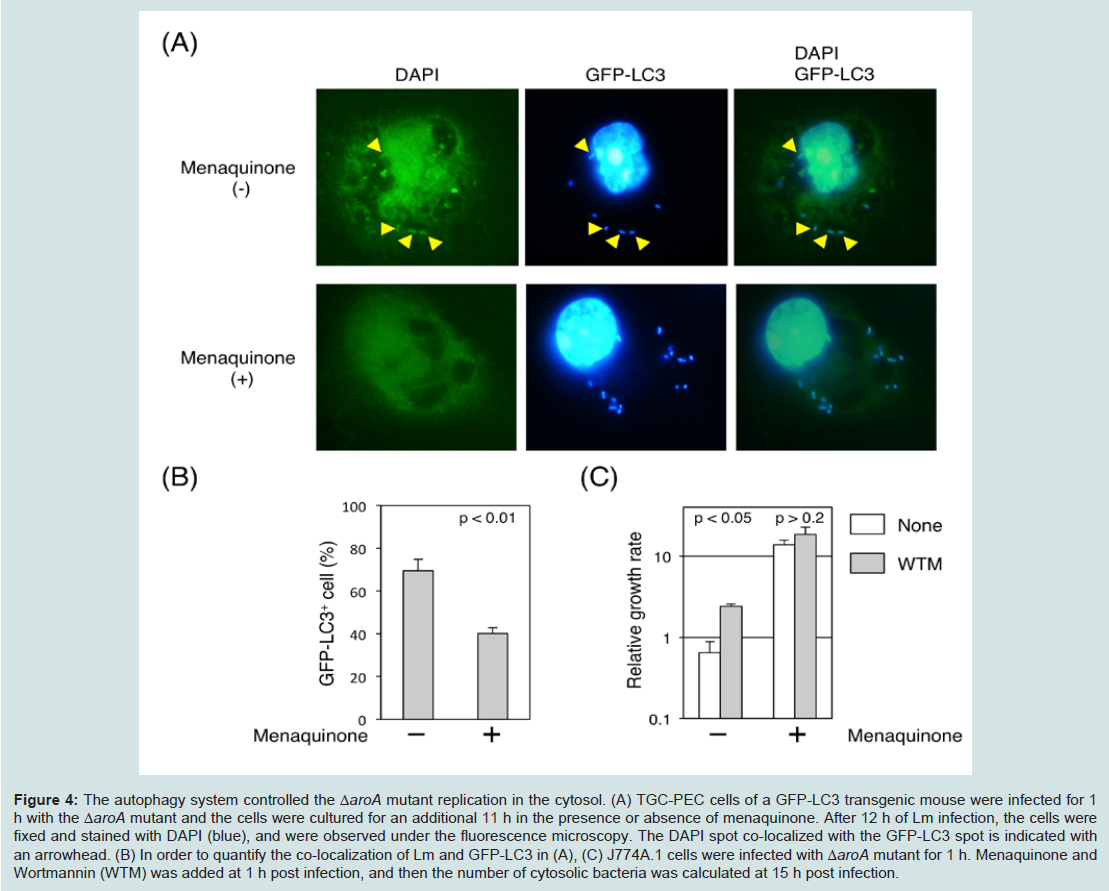 Figure 4: The autophagy system controlled the ΔaroA mutant replication in the cytosol. (A) TGC-PEC cells of a GFP-LC3 transgenic mouse were infected for 1 h with the ΔaroA mutant and the cells were cultured for an additional 11 h in the presence or absence of menaquinone. After 12 h of Lm infection, the cells were fixed and stained with DAPI (blue), and were observed under the fluorescence microscopy. The DAPI spot co-localized with the GFP-LC3 spot is indicated with an arrowhead. (B) In order to quantify the co-localization of Lm and GFP-LC3 in (A), (C) J774A.1 cells were infected with ΔaroA mutant for 1 h. Menaquinone and Wortmannin (WTM) was added at 1 h post infection, and then the number of cytosolic bacteria was calculated at 15 h post infection.
Figure 4: The autophagy system controlled the ΔaroA mutant replication in the cytosol. (A) TGC-PEC cells of a GFP-LC3 transgenic mouse were infected for 1 h with the ΔaroA mutant and the cells were cultured for an additional 11 h in the presence or absence of menaquinone. After 12 h of Lm infection, the cells were fixed and stained with DAPI (blue), and were observed under the fluorescence microscopy. The DAPI spot co-localized with the GFP-LC3 spot is indicated with an arrowhead. (B) In order to quantify the co-localization of Lm and GFP-LC3 in (A), (C) J774A.1 cells were infected with ΔaroA mutant for 1 h. Menaquinone and Wortmannin (WTM) was added at 1 h post infection, and then the number of cytosolic bacteria was calculated at 15 h post infection.Discussion
It has been established that Lm induces cell death in macrophages, hepatocytes and dendritic cells [4,5,8]. It is accepted that the cell death of Lm-infected macrophages is induced by LLO, which has a crucial role in the escape of the bacteria from the vacuole [7]. To evaluate the requirement of LLO for inducing cell death, we used an hlyW492A mutant, which possesses a mutation in codon Trp-492 of LLO that leads to decreased hemolytic activity in vitro [20]. Despite this 99.9% reduction in hemolytic activity, the hlyW492A mutant mutant can escape from the phagosome into the cytosol, and induce the death of macrophages. This may indicate that activity of LLO is not essential for the induction of cell death. This idea is further supported by our finding that the ΔaroA mutant, which has normal LLO activity and reduced replication potential in the cytosol because of a defect in oxidative metabolism [21], failed to induce cell death in macrophages. These results indicate that LLO is necessary for Lm to invade the cytosol, but is not sufficient to induce Lm-infected cell death in macrophages.In our study comparing the intracellular Lm growth and the induction of cell death in macrophages, we noticed that the induction of cell death correlated with the intracytosolic Lm growth. The mutant possessing a low replication rate in the cytosol (ΔaroA) appeared to be surrounded by the autophagosome, suggesting the involvement of an autophagic mechanism in rescuing the host cells from Lm-induced cell death when the Lm grow slowly in the cytosol. WT Lm, which has a high replication rate, appears to be able to escape from autophagic control. Menaquinone is an essential component of the respiratory chain in Lm, and a defect in menaquinone biosynthesis is expected to impair oxidative phosphorylation [21]. The ΔaroA mutant with normal LLO activity escapesfrom the phagosome into the cytosol, but grows slowly in the cytosol because of the deficiency of menaquinone. Although the ActA protein prevents the bacteria from being surrounded by the autophagosome [18], the ΔaroA mutant with both the ActA and LLO proteins still is surrounded by the LC3-positive membrane and destroyed. Therefore, when Lm grows slowly in the cytosol, even in the presence of LLO and ActA, the autophagy system rescues the host cells from infection-induced cell death. In the case of WT Lm, the intermediate substance of oxidative metabolism or the rapid replication of the Lm might interrupt the autophagy machinery and induces death in the host cells. However, the precise mechanism that activates or permits the activation of the autophagy machinery in ΔaroA mutant-infected macrophages has not yet been determined.
Because of the nature of its growth in host cells, Lm is thought to be a candidate vector for delivering plasmids into the cytosol [26-28]. However, there have been problems associated with the virulence of Lm that have prevented its use. Since the ΔaroA mutant used here has low virulence and low toxicity to host cells, it may be a good candidate vector for applications in immunotherapy.
Acknowledgements
We thank Dr. P. Cossart for providing us with the WT and mutant Listeria. We also thank Dr. H Shinomiya for his valuable support and helpful discussions. This work was supported in part by a grant in aid from the Ministry of Education, Culture, Sports, Science andTechnology of Japan, and by Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (B) Grant 23390075.References
- Pamer EG (2004) Immune responses to Listeria monocytogenes. Nat Rev Immunol 4: 812-823.
- Kaufmann SH (1993) Immunity to intracellular bacteria. Annu Rev Immunol 11: 129-163.
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, et al. (2001) Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14: 584-640.
- Cossart P, Lecuit M (1998) Interactions of Listeria monocytogenes with mammalian cells during entry and actin-based movement: bacterial factors, cellular ligands and signaling. EMBO J 17: 3797-3806.
- Gaillard JL, Berche P, Sansonetti P (1986) Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect Immun 52: 50-55.
- Pizarro-Cerda J, Cossart P (2006) Subversion of cellular functions by Listeria monocytogenes. J Pathol 208: 215-223.
- Guzman CA, Domann E, Rohde M, Bruder D, Darji A, et al. (1996) Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol 20: 119-126.
- Cossart P, Pizarro-Cerda J, Lecuit M (2003) Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol 13: 23-31.
- Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907-1916.
- Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99-109.
- Orvedahl A, Levine B (2009) Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 16: 57-69.
- Ogawa M, Mimuro H, Yoshikawa Y, Ashida H, Sasakawa C (2011) Manipulation of autophagy by bacteria for their own benefit. Microbiol Immunol 55: 459-471.
- Levine B, Kroemer G (2009) Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ 16: 1-2.
- Yoshimori T (2004) Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun 313: 453-458.
- Rich KA, Burkett C, Webster P (2003) Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol 5: 455-468.
- Py BF, Lipinski MM, Yuan J (2007) Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3: 117-125.
- Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, et al. (2007) Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3: 442-451.
- Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, et al. (2009) Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11: 1233-1240.
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, et al. (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032-1036.
- Michel E, Reich KA, Favier R, Berche P, Cossart P (1990) Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid substitutions in listeriolysin O. Mol Microbiol 4: 2167-2178.
- Stritzker J, Janda J, Schoen C, Taupp M, Pilgrim S, et al. (2004) Growth, virulence, and immunogenicity of Listeria monocytogenes aro mutants. Infect Immun 72: 5622-5629.
- Stritzker J, Schoen C, Goebel W (2005) Enhanced synthesis of internalin A in aro mutants of Listeria monocytogenes indicates posttranscriptional control of the inlAB mRNA. J Bacteriol 187: 2836-2845.
- Levine B (2005) Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120: 159-162.
- Levine B, Deretic V (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 7: 767-777.
- Munz C (2009) Enhancing immunity through autophagy. Annu Rev Immunol 27: 423-449.
- Shen H, Tato CM, Fan X (1998) Listeria monocytogenes as a probe to study cell-mediated immunity. Curr Opin Immunol 10: 450-458.
- Shen H, Kanoh M, Liu F, Maruyama S, Asano Y (2004) Modulation of the immune system by Listeria monocytogenes-mediated gene transfer into mammalian cells. Microbiol Immunol 48: 329-337.
- Shen H, Kanoh M, Maruyama S, Matsumoto A, Zhang W, et al. (2008) Attenuated Listeria-infection activates natural killer cell cytotoxicity to regress melanoma growth in vivo. Microbiol Immunol 52: 107-117.