Research Article Regulation of Adherens Junctions in Trabecular Meshwork Cells by Rac GTPase and their influence on Intraocular Pressure
Padmanabhan P Pattabiraman1, David L Epstein1 and Ponugoti Vasantha Rao1,2*
- 1Department of Ophthalmology, Duke University School of Medicine, Durham, NC, USA 27710
- 2Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, NC, USA 27710
*Address for Correspondence: Ponugoti Vasantha Rao, Ph.D, Department of Ophthalmology, Duke University School of Medicine, Durham, NC, USA 27710, Tel: 919-681-5883; Fax: 919-684-8983; E-mail:
rao00011@mc.duke.edu Citation: Pattabiraman PP, Epstein DL, Vasantha Rao P. Regulation of Adherens Junctions in Trabecular Meshwork Cells by Rac GTPase and their Influence on Intraocular Pressure. J Ocular Biol. 2013;1(1): 10.
Copyright © 2013 Pattabiraman PP, et al. This is an open access article distributed under the
Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 1, Issue: 1
Submission: 26 June 2013 | Accepted: 01 July 2013 | Published: 05 July 2013
Abstract
Intercellular adherens junctions and cell-extracellular matrix interactions are presumed to influence aqueous humor (AH) drainage via the conventional route, however, their direct role in modulation of intraocular pressure (IOP) is not well understood. Here, we investigated the role of Rac GTPase signaling in basal and growth factor-induced formation of adherens junctions in human trabecular meshwork (HTM) cells as compared to human umbilical vascular endothelial cells, and evaluated the effects of inhibition of Rac GTPase activity on IOP in rabbits. Expression of a constitutively active Rac1 GTPase or treatment with platelet derived growth factor (PDGF), a known activator of Rac GTPase, induced formation of β-catenin-based adherens junctions, actin cytoskeletal reorganization and membrane ruffle in HTM cells. In contrast, treatment of HTM cells with inhibitors of Rac GTPase caused cell-cell separation, a decrease in adherens junctions, and reorganization of actin stress fibers to the cell cortical regions and focal adhesion to the cell leading edges. Both, constitutively active Rac1 and PDGF stimulated generation of Reactive Oxygen Species (ROS) in HTM cells, and ROS were found to increase adherens junction formation and transendothelial electrical resistance (TEER) in HTM cells. Topical application of Rac GTPase inhibitors (EHT1864 and NSC23766), however, only marginally influenced IOP in rabbit eyes. Taken together, these data reveal that while Rac GTPase signaling plays a significant role in regulation of adherens junctions, ROS production and TEER in cells of the AH outflow pathway, Rac inhibitors showed only a marginal influence on IOP in live rabbits.
Keywords
Rac GTPase; Adherens junctions; Cytoskeleton; Trabecular meshwork; ROS; Intraocular pressure
Introduction
Glaucoma is the second leading cause of blindness in the United States. A major risk factor for primary open-angle glaucoma is elevated intraocular pressure (IOP) caused by increased resistance to aqueous humor (AH) outflow localized within the conventional/trabecular pathway [
1-3]. Dysregulated transendothelial permeability barrier activity of Schlemm’s canal stemming from altered cell-cell and cell-extracellular matrix (ECM) interactions is presumed to be partly responsible for increased resistance to drainage of AH through the conventional pathway [
4-8]. Extracellular cues such as growth factors, ECM and steroids are thought to influence AH outflow resistance via altering the cytoskeletal organization, contractile properties, cell-cell junctions, cell-ECM interactions and permeability barrier of cells within the AH outflow pathway [
4,7,9-15]. The mechanistic basis by which cell-cell junctions in AH outflow pathway are regulated, and the direct role of these complexes in AH drainage and IOP homeostasis, however, has remained elusive. Therefore, studies to address the formation and regulation of the adherens junctions, which represent a predominant type of cell-cell junction in endothelia [
16], seem both necessary and critical for exploring a potential role for these junctions in IOP homeostasis. This knowledge may provide important insights into the etiology of glaucoma and support development of novel therapies.
The conventional AH drainage pathway consists of the trabecular meshwork (TM), juxtacanalicular tissue (JCT) and Schlemm’s canal (SC) [
3]. The TM is a filter made up of collagen-based connective tissue organized into a network of beams covered by endothelial-like trabecular cells. As AH moves from the TM to the JCT adjacent to the inner wall of SC, the spaces between the beams decrease [
3,17]. Although the maximal resistance to AH outflow is thought to occur at the junction where the TM meets the inner wall of SC [
17,18], it is also commonly believed that the TM, SC and JCT contribute to resistance to AH drainage via maintenance of normal cell shape, cell-cell junctions and cell-ECM interactions, which subsequently influence permeability barrier integrity and tissue stiffness, analogous to what occurs in the vascular endothelium [
12,18,19-21]. Further, it is well documented that the cells of the conventional outflow pathway possess endothelial-like properties [
20,22]. The endothelium acts as a semi-permeable barrier and regulates fluid and solute exchange in many different tissue types [
23-25]. Permeability of endothelial cells is determined by the adhesive properties of the proteins that comprise the tight junctions and adherens junctions [
24-28].
Currently there are a few published studies of the AH outflow pathway that directly address the importance of cell-cell junctions and their role in modulation of AH drainage and IOP [
4,6,7,14,15,21,29]. Syriani et al. [
30] recently reported that perfusion of PDGF through enucleated bovine eyes increases AH outflow facility likely through the stimulation of Rac GTPase activity and altered cytoarchitecture and cell-cell junctions in cells of the outflow pathway. However, other agents which are known to activate Rac GTPase activity and stimulate cell-cell junction formation including Sphingosine-1-phosphate, have been shown to decrease AH outflow facility in perfused model systems possibly via increased transendothelial permeability barrier of SC [
7,10,31]. However, in these latter studies, the direct role of Rac GTPase activity in either cell-cell junction formation or on IOP has not been addressed.
The Rho GTPases including Rac and Rho, have been demonstrated to regulate transendothelial permeability barrier by controlling actin cytoskeletal reorganization, contractile properties, cell-cell junctions and cell-ECM interactions in different cell types [
26,28,32].
Moreover, the aberrant activity of Rho GTPases underlying impaired permeability barrier function in endothelial cells is known to be involved in various pathological conditions [
24,33,34]. Various growth factors, mechanical stress and oxidative stress are also recognized to influence activity of the Rho GTPases and transendothelial permeability [
24,28,29,35]. Therefore, in this study, we evaluated the direct effects of Rac GTPase activation and inactivation on adherens junctions formation in HTM cells as compared to human umbilical vascular endothelial cells (HUVEC) used as a model SC endothelial cell type, and on IOP changes in a live animal model.
Materials and Methods
Reagents
Two different Rac inhibitors- NSC23766 (N6-[2-[[4-(Diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine trihydrochloride) and EHT1864(5-[5-[7-(Trifluoromethyl)quinolin-4-ylthio]pentyloxy]-2-(morpholinomethyl)-4H-pyran-4-one dihydrochloride) that inhibit either Rac GTPase interaction with guanosine nucleotide exchange factors [
36] or Rac GTPase activation [
37,38], respectively, were purchased from Tocris Biosciences (Ellisville, MO) and Sigma-Aldrich St. Louis, MO), respectively. Rho kinase inhibitor (Y27632) was provided by Welfide Corp (Tokyo, Japan). Human recombinant PDGF AA (CYT-341) was purchased from Prospec (Israel). N-acetyl-L-cysteine (A7250), hydrogen peroxide solution (H1009), diphenyleneiodonium chloride (D2926), mouse monoclonal anti-vinculin (V4505), anti-tubulin (B5168), anti-β-catenin (C7207), rhodamine conjugated phalloidin and FITC conjugated phalloidin were purchased from Sigma-Aldrich (St. Louis, MO). 2′,7′-dichlorodihydrofluorescein diacetate was purchased from Invitrogen (Grand Island, NY).
Mouse monoclonal anti- VE-Cadherin antibody (MAB1989) was procured from EMD Millipore (Billerica, MA). Phospho-myosin light chain antibody (36745) was from Cell Signalling Technology (Danvers, MA).
HTM and HUVEC cell cultures
Human TM (HTM) cells were cultured from the TM tissue derived from donor eyes (obtained from local eye banks) as we described earlier [
39]. Cell cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and penicillin (100 U/500 ml)-streptomycin (100 mg/500 ml)-glutamine (4 mM). Human umbilical vascular endothelial cell line (HUVEC) purchased from Clonetics (San Diego, CA) was cultured in EGM-2 (Endothelial Growth Medium) medium supplemented with FBS, hydrocortisone, rhEGF, bovine brain extract and antibiotics (Gentamicin sulphate and Amphotericin-B) as per the manufacturer’s instructions (Clonetics). All cells were cultured at 37° C under 5% CO
2. HTM cells were used between passages 4-6 and HUVECs were used between passages 7-9. HUVEC cells were used as a model SC endothelial cell type for the purpose of understanding the regulation of adherens junctions and permeability characteristics.
Immunofluorescence staining and microscopy
HTM and HUVEC cells were grown on gelatin (2%)-coated glass coverslips until they attained confluency. For HTM cells, after appropriate treatments, cells were washed with phosphate buffered saline (PBS) twice and fixed in 4% paraformaldehyde/PBS for 15 min. Coverslips were then washed thrice in 1xPBS, cells permeabilized in 1% Triton X-100 in 1x PBS (PBST buffer) and blocked with serum buffer containing 5% FBS in PBST buffer for 60 min. The coverslips were incubated with the respective primary antibodies for 2 h at room temperature under humidity, and rinsed with permeabilization buffer followed by 3 washes with serum-containing buffer. The coverslips were then incubated with Alexa fluor 480 or 594-conjugated secondary antibodies in serum/PBS buffer for 2 h at room temperature and after three washes with 1x PBS, mounted onto glass slides with Aqua Mount (Lerner Laboratories, Pittsburg, PA) [
39]. HUVEC cells were washed twice in Hanks Buffered Salt Solution (HBSS) following treatment with the appropriate drug, fixed in 4% paraformaldehyde in HBSS for 20 min at room temperature. Cells were subsequently washed once with 0.1 M glycine in HBSS for 5 min and three washes with HBSS for 10 min. Blocking (1% Triton X-100 and 5% FBS in HBSS) was done for 1 h at room temperature, after which cells were incubated with the respective primary antibodies and Alexa fluor-conjugated secondary antibodies for 2 h at room temperature. Later, the coverslips were washed thrice with HBSS and mounted onto glass slides as described above. The slides were observed under a Nikon confocal system (C1 Digital Eclipse), and z-stack images were collected and processed.
Adenovirus-mediated gene transduction
Replication defective recombinant adenoviral vectors encoding either LacZ alone or constitutively active Rac1 (Rac1G12V) and dominant negative Rac1 (Rac1T17N) were obtained from the Toren Finkel laboratory, National Heart Lung and Blood Institute, NIH [
40,41]. The viral vectors were amplified and purified using the Adeno-x Maxi Purification kit (Clontech) as we described earlier [
11]. HTM cells grown either on gelatin-coated glass coverslips or in plastic petridishes to 80-85% confluency were infected with adenovirus at a multiplicity of infection (MOI) of approximately 70, as titrated using the Adeno-X Rapid titer kit (Clontech). After achieving more than 80% transfection efficiency (assessed based on LacZ immunostaining or Myc staining for Rac1G12V and Rac1T17N) at 36 h post-infection, cells were serum starved or maintained in low serum (1%) media for 24 h before being used in the respective experiments.
Myosin light chain phosphorylation
The effect of Rac inhibition by EHT1864 and Rac activation by PDGF on phosphorylation status of myosin light chain (MLC) in confluent cultures of HTM cells (serum starved for 24 h) was determined by urea-glycerol gel electrophoresis as we described earlier [
39]. Briefly, confluent cell cultures were extracted with cold 10% trichloroacetic acid following treatment with PDGF or Rac inhibitors and cell precipitates collected after centrifugation at 13,000 rpm were washed and finally dissolved in 8M Urea buffer by sonication. 20 μg of protein was separated on urea-glycerol gels and transferred onto nitrocellulose filters. The filters were then subjected to immunoblot analysis using anti-phospho MLC2 antibody and blots were developed by enhanced chemiluminescence (ECL). Blots were scanned with Image J software (NIH) to quantify the differences in immunopositive band densities.
Measurement of intracellular Reactive Oxygen Species (ROS) production
Intracellular ROS production was quantified with the cellpermeant ROS indicator 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) [
42]. Briefly, treated and control confluent HTM cells were trypsinized and incubated in 1 ml PBS containing 20 μM H2DCFDA for 30 min. After this loading period, the cells were washed with PBS, and the mean green fluorescence of 10,000 cells was immediately recorded and quantified by flow cytometry (BD FACSCalibur
TM) and the system software (FITC/FL-1 channel; CellQuest; BD Biosciences). Unstained control cells were included to evaluate baseline fluorescence. The laser intensity settings were adjusted to the lowest level at which autofluorescence was not detected.
H2O2 and hyperoxia treatments
HTM cell cultures (passage 4) were subjected to H
2O
2 treatment or hyperoxia conditions. For H
2O
2 treatment, confluent cultures of HTM cells were maintained in DMEM containing 1% FBS for 24 h, then treated with 250 μM of H
2O
2 for 3 h alone or in the presence of DPI (2 μM) or NAC (1 mM). For subjecting the HTM cell cultures to normobaric hyperoxia conditions, confluent cultures of HTM cells were grown for 7 days at 40% O
2 and 5% CO
2 in DMEM containing 1% FBS. On the other hand, the control cultures were grown under physiological oxygen conditions (5% O
2 and 5% CO
2) in a triple-gas incubator as described by Liton et al. [
43]. These treatments were carried out in the presence and absence of DPI (2 μM) or NAC (1 mM).
Transendothelial electrical resistance (TEER) measurement
HTM cells (donor age 77, 64 and 55 years) were plated at confluence on polycarbonate Snapwell permeable supports (12 mm Snapwell insert with 0.4 m pore polycarbonate membrane #3407, Costar) and maintained in DMEM supplemented with 10% FBS until reaching a net TEER of at least 5 Ohm
*cm
2 (by 5-7 days). Net TEER refers to the difference in the resistance of membrane containing confluent monolayer of cells compared to membrane alone. HTM cells were then maintained in DMEM containing 1% FBS or switched to DMEM/1% FBS containing the test compounds [Rac inhibitor NSC23766 (50 μM), DPI (2 μM), NAC (1 mM) or PDGF (10 ng/ml)] at their respective concentrations. In a separate experiment, PDGF was added after 6 h of pretreatment with either DPI or NAC. TEER measurements were recorded using an Endohm device (World Precision Instruments) as described by Perkumas and Stamer [
44]. The net TEER was calculated by averaging the resistance collected from three independent Snapwell membranes for each treatment.
IOP measurements in rabbits
Experiments were performed on 6 healthy adult Dutch-Belted male rabbits weighing around 5 lb adhering to the NIH statement for the Use of Animals in Biomedical Research with approved protocol by the Duke IACUC. Intraocular pressure (IOP) was recorded using a pneumotonometer after topical application of 0.05% proparacaine as we described earlier [
45]. One eye was treated with vehicle (PBS) and the other eye was treated with drug (50 mM of NSC23766 or 25 mM of EHT1864) prepared in PBS. The drug was applied as two 30 μl drops with a one minute interval, and with the animal’s eye lids being kept open during drug application to prevent blinking. Baseline pressure was recorded just before the application of the drug and IOP was recorded at 1, 2, 4, 6 and 24 h post drug application. IOP changes between the test and control eyes at different time points, and changes between the baseline IOP and drug treated IOP were evaluated using appropriate statistical tests.
Analysis of drug levels in aqueous humor
The concentration of NSC23766 in rabbit AH was measured 1 h post topical dosing of drug (50 mM). Briefly, after 1 h of topical application of drug, animals were anesthetized by injecting them subcutaneously with 0.5 ml ketamine/xylazine, followed by a wait of 3 to 5 min, and euthanization using 2 ml of pentobarbital. Immediately following this procedure, the AH was collected by a board certified ophthalmologist using a 29 gauge needle syringe. The AH specimens were frozen until further analysis. Drug concentration in AH specimens was quantified by liquid chromatography-mass spectrometry (LC/MS) analysis [
46]. These analyses were done at the Scripps Research Institute, Florida (with the generous help of Dr. Philip Lograsso). Known concentrations of drug dissolved in PBS were used as standards to determine the drug concentration in the AH specimens. Data represent the average of two independent specimens. The lower limit of quantification for the LC/MS method used was 5 nM.
Statistical analysis
Student’s t-test (for cell-based assays) and Wilcoxon Rank-Sum test (for animal studies) were applied to analyze statistical significance. Minimum values of P < 0.05 were considered as statistically significant.
Results
Inhibition of Rac GTPase alters cell shape and actin cytoskeletal organization and decreases adherens junctions in HTM and HUVEC cells
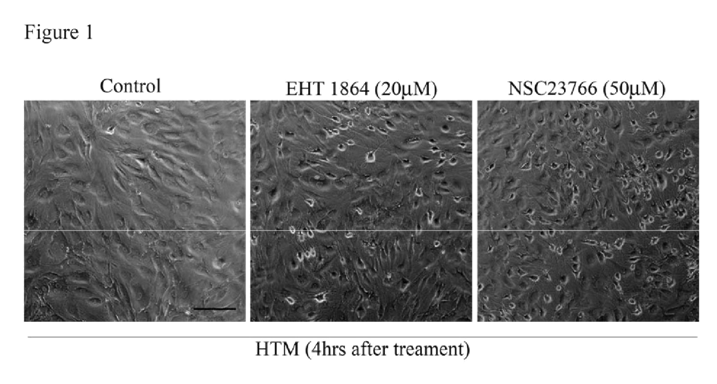
To understand the role of Rac GTPase signaling in regulation of actin cytoskeleton reorganization and adherens junctions formation in TM cells, we first determined the effects of Rac inhibitors on these processes. Confluent cultures of HTM cells maintained overnight in 1% FBS were treated with Rac GTPase inhibitors- EHT1864 or NSC23766 (1 M to 50 μM; dissolved in PBS) for 6 h. After 4 h of exposure to EHT1864 (20 μM) or NSC23766 (50 μM), cells exhibited a contractile morphology, as assessed by phase contrast imaging, and relative to untreated control cells (
Figure 1). There were no detectable changes in cell morphology at lower concentrations of drug (data not shown). The notable feature was that drug treated cells lost contact from adjacent cells but did not detach from the surface. These drug-induced morphological changes were found to be reversible upon drug washout with PBS, with cells recovering a normal morphology by 24 h following drug removal. These drugs did not cause any notable cytotoxicity based on live cell labeling for the enzymatic hydrolysis of fluorescein diacetate and nuclear labeling using propidium iodide staining (data not shown); [
39].
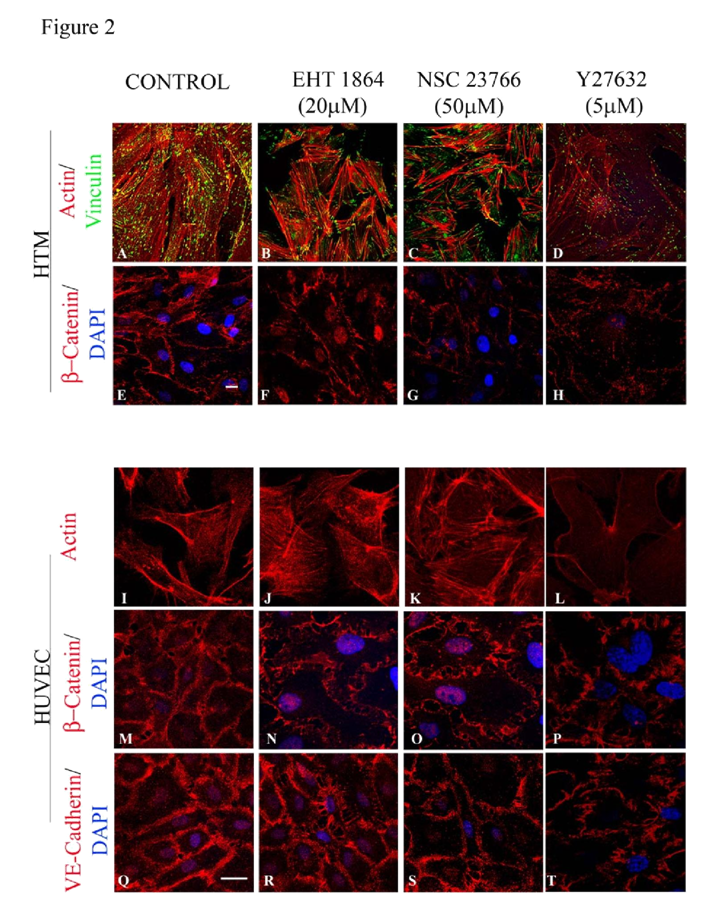
Treatment of confluent cultures of HTM cells maintained in 1% FBS with EHT1864 (20 μM) or NSC23766 (50 μM) for 4 h induced increased assembly of actin stress fibers at the cortical regions, confirmed by staining for F-actin (Rhodamine-Phalloidin) (
Figure 2B and 2C) in both HTM (
Figure 2B and 2C) and HUVEC (
Figure 2J and 2K) cells, and increased focal adhesions formation (vinculin staining; green staining) especially at the leading edges (
Figure 2B and 2C) in HTM cells, compared to corresponding controls (
Figure 2A and 2I). β-catenin and VE-cadherin, components of adherens junction complexes, were immunostained in confluent cultures of both HTM and HUVEC cells. HTM cells exhibited a well-organized positive staining pattern/profile for β-catenin (
Figure 2E), but not for VEcadherin, while the HUVEC cells stained positively for both proteins (
Figure 2M and 2Q), which were found to localize at the cell-cell contacts. Treatment with EHT1864 (20 μM) or NSC23766 (50 μM) for 4 h led to a decrease in β-catenin staining in HTM cells (
Figure 2F and 2G), and decreased staining of both β-catenin (
Figure 2N and 2O) and VE-cadherin (
Figure 2R and 2S) in HUVEC cells, with disjointed cell-cell junctions compared to the controls (
Figure 2E, 2M and 2Q). In contrast, inhibition of Rho kinase by Y27632 (5 μM for 4 h), led to a loss of stress fibers and focal adhesions as well as adherens junctions in both HTM (
Figure 2D and 2H) and HUVEC (
Figure 2L, 2P and 2T) cells.
Activation of Rac GTPase by PDGF induces adherens junctions in HTM and HUVEC cells
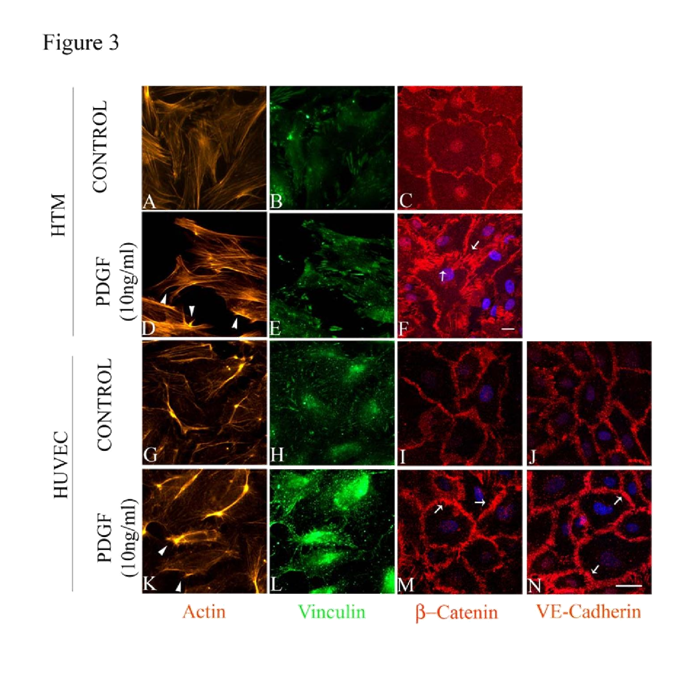
PDGF has been demonstrated to activate Rac GTPase in various cell types including TM cells [
30,
47,48]. Serum starved (for 36 h) HTM and HUVEC cells treated with 10 ng/ml PDGF AA (dissolved in PBS) exhibited subtle morphological changes including membrane ruffle and lamellipodia-like structures based on F-actin (Rhodaminephalloidin) staining by 1 h (
Figure 3D and 3K, arrow heads) compared to controls (
Figure 3A and 3G). Similarly, focal adhesions (based on vinculin staining in green) showed only a subtle reorganization (
Figure 3E and 3L) in the PDGF treated cells compared to the controls (
Figure 3B and 3H). Under similar treatment conditions, we examined the effects of PDGF on distribution of β-Catenin and VE-Cadherin by immunofluorescence labeling. In HTM cells, β-Catenin staining was found to be more prominent at the cell-cell junctions under PDGF stimulation (
Figure 3F) compared to the control (
Figure 3C). In unstimulated HUVEC cells, β-Catenin atenin and VE-Cadherin staining was seen along the cell-cell contact area (
Figure 3I and 3J, respectively) but upon stimulation with PDGF (10 ng/ml for 2 h), the staining intensity of these molecules was found to be much stronger compared to the controls (
Figure 3M and 3N). Under these conditions, the cell shape changes were found to be not obviously different between the treated and control cells based on phase contrast microscope imaging (not shown).
Effects of constitutively active and dominant negative forms of Rac1 GTPase on adherens junctions in HTM and HUVEC cells
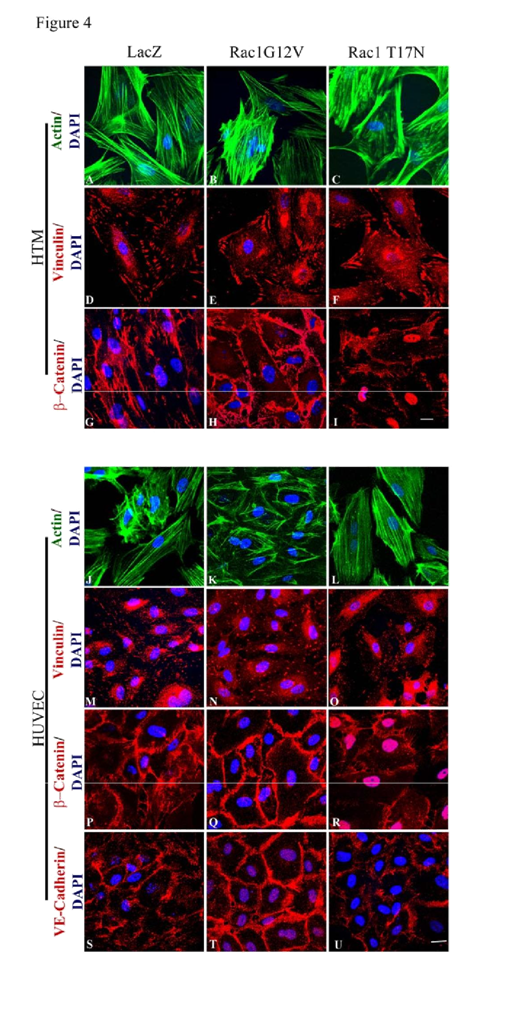
In order to assess the role of Rac GTPase signaling on the properties of adherens junctions in HTM and HUVEC cells, these cells were infected (80% confluent) with the adenovirus (at 70 MOI) expressing either the constitutively active (Rac1G12V) or dominant negative (Rac1T17N) forms of Rac GTPase, or a LacZ control. Infection efficiency was evaluated based on β-galactosidase expression encoded by the LacZ or
myc gene expression by immunoblotting (data not shown) or by immunofluorescence labeling. By around 36 h post infection, nearly 80% of the cells expressed detectable levels of β-galactosidase or
myc protein. The cells infected with Rac1G12V were serum starved for 24 h (after 36 h of virus infection), while the cellsinfected with Rac1T17N or LacZ vectors were maintained in 1% FBS media. The LacZ expressing HTM and HUVEC cells revealed no changes in either actin cytoskeleton organization or cell-cell junctions compared to their respective non-infected controls (data not shown). The Rac1G12V expressing HTM (
Figure 4E) and HUVEC (
Figure 4N) cells exhibited only subtle changes in focal adhesions organization but stronger β-catenin (in both HTM and HUVEC cells;
Figure 4H and 4Q) and VE-Cadherin (in HUVEC cells;
Figure 4T) staining at the cell-cell junctions compared to the LacZ controls (
Figure 4D, 4G, 4M, 4P and 4S). In contrast, the Rac1T17N expressing HTM (
Figure 4C and 4F) and HUVEC (
Figure 4L and 4O) cells displayed strong cortical actin staining with much reduced and discontinuous staining of β-catenin (in both HTM and HUVEC cells;
Figure 4I and 4R) and VE-cadherin (in HUVEC cells;
Figure 4U), indicating loss of cell-cell junctions in response to overexpression of the dominant negative form of Rac1 in both HTM and HUVEC cells. Under above described conditions, the cell shape of either HTM or HUVEC cells exhibited no marked differences from the respective control cells based on phase contrast microscope examination (data not shown).
Semi-confluent HTM and HUVEC cell cultures were infected with adenovirus expressing the LacZ as control (LacZ), or constitutively active Rac (Rac1G12V) or dominant negative Rac (Rac1T17N). Confluent cell cultures were serum starved for 24 h after virus infection in the case of Rac1G12V or maintained in 1% serum in the case of LacZ and Rac1T17N. Following this, the cells were fixed and immunostained for F-actin, vinculin, β-catenin and VE-cadherin. The Rac1G12V expressing cells showed only subtle changes in actin stress fibers (FITC-phalloidin; green) and focal adhesions (vinculin staining; red) in both HTM and HUVEC cells (B, E, K, N). Similar to Rac inhibitors, actin stress fibers in the Rac1T17N expressing cells exhibited their reorganization to the cortical regions intensely and focal adhesions to the leading edges (C, F, L, O) compared to the control cells (A, D, J, M). Intense and increased immunostaining for β-catenin (in HTM and HUVEC cells) and VE-cadherin (in HUVEC cells) was observed in the Rac1G12V transduced cells (H, Q, T) while Rac1T17N transduced cells showed increased intercellular gaps and decreased β-catenin (I, R, in HTM cells) and VE-cadherin (U, in HUVEC cells) compared to the LacZ controls (G, P, S). Scale bar: 10 μm.
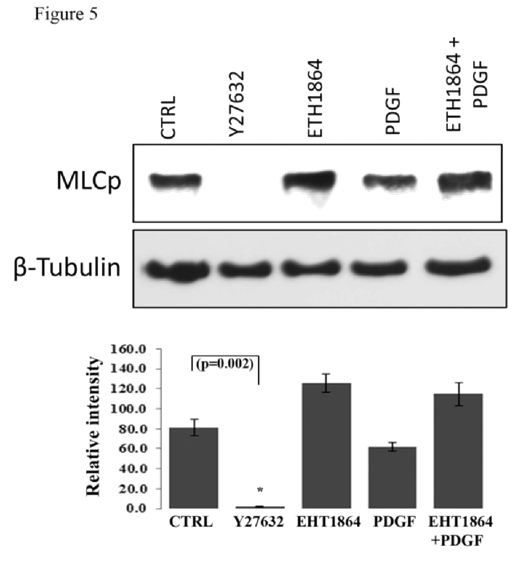
Influence of Rac GTPase inhibition on MLC phosphorylation in HTM cells
Myosin light chain (MLC), which is the regulatory subunit of myosin II, is phosphorylated by myosin light chain kinase (MLCK) and Rho kinase, and phosphorylation of MLC controls actin-myosin II cross-bridging, leading to cellular contraction [
49,50]. Here we tested the effects of Rac GTPase activity on MLC phosphorylation in HTM cells (
Figure 5). Treatment of confluent cultures of HTM cells maintained in 1% FBS (overnight) with EHT1864 (20 μM for 2 h) led to an increase, albeit insignificant, in the levels of MLC phosphorylation compared to the respective controls. On the other hand, stimulation of serum starved HTM cells with human recombinant PDGF (10 ng/ml) caused a slight decrease in MLC phosphorylation, while treatment with Rho Kinase inhibitor- Y27632 (5 μM) for 2 h led to a dramatic and significant (P < 0.05; n=3) decrease in the levels of MLC phosphorylation, as has been reported earlier [
15]. PDGF stimulation which is expected to activate Rac GTPase activity in human TM cells [
30] pretreated with Rac inhibitor (EHT1864 for 4 h) did not lower the levels of MLC phosphorylation (
Figure 5). β-Tubulin was immunobloted for loading normalization.
Confluent cultures of HTM cells were maintained in 1% FBS overnight and treated with the Rac GTPase inhibitor- EHT1864 (20 μM) or Rho kinase inhibitor (Y27632, 5 μM). A second set of cells under serum free conditions was treated with PDGF (10 ng/ml) for 4 h. Rac GTPase inhibitor and PDGF exerted contrasting but moderate effects on MLC phosphorylation, with Rac inhibitor activating and PDGF inhibiting the response, compared to the Rho kinase inhibitor, which caused a complete suppression of MLC phosphorylation (P < 0.05; n=3). Activation of Rac GTPase by PDGF (10 ng/ml for 2 h, in serum free media) after pretreatment with the Rac inhibitor EHT1864 (20 μM in 1% serum media for 4 h) did not cause any additional changes in the levels of myosin light chain phosphorylation indicating that PDGF induced changes in MLC phosphorylation appear to be mediated through the Rac GTPase. β-Tubulin immunoblot was used for loading control. Histograms show quantitative changes in MLC phosphorylation based on densitometric analysis using Image J. Error bars represent standard error. N=3.
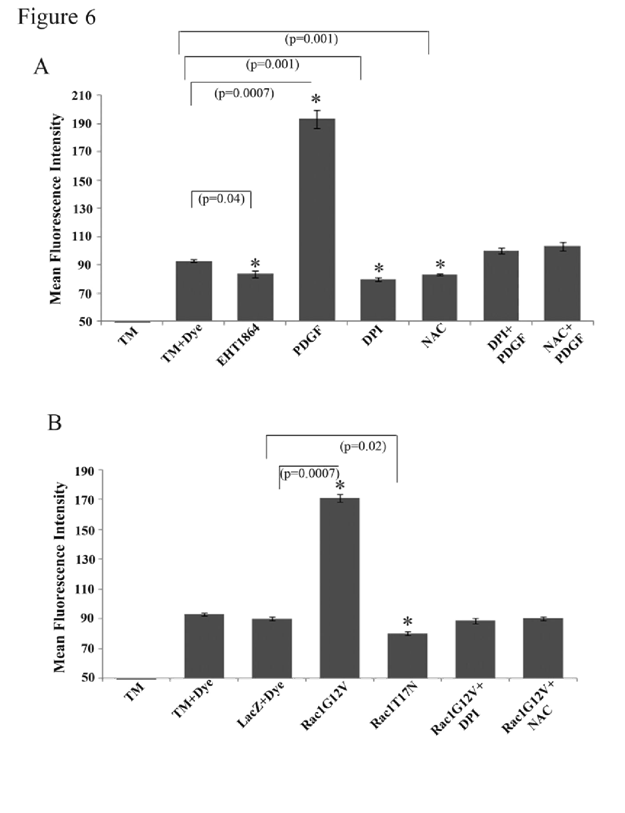
Formation of Rac GTPase-mediated adherens junctions is dependent on ROS production in HTM cells
Rac GTPase signaling has been shown to regulate the generation of ROS by activating the NADPH oxidase system [
51,52]. To explore whether ROS are generated and in turn influence cell-cell interactions in HTM cells, confluent cultures (maintained under serum free conditions) were stimulated either with PDGF (10 ng/ml for 4 h) or by overexpression of Rac1G12V (for nearly 48 h), followed by determination of ROS levels by flow cytometry using the H2DCFDA substrate. There was a significant increase (nearly two fold; p < 0.05) in the amount of ROS production under both conditions known to promote Rac activation (i.e., PDGF stimulation and overexpression of Rac1G12V,
Figure 6A and 6B, respectively). Presence of Rac inhibitor EHT1864 (20 μM for 4 h) or overexpression of dominant negative Rac (RacT17N) for 48 h (in 1% serum) per se caused a significant (~10 and 14%, respectively) decrease in ROS levels, compared to the basal ROS levels in LacZ transfected or untreated control cells (
Figure 6A and 6B). Quenching of ROS by NAC (1 mM N-acetyl cysteine for 2 h; dissolved in PBS) or inhibition of the NADPH oxidase system by DPI- (2 μM diphenyleneiodonium for 2 h; dissolved in DMSO) led to a significant (~11 and 15%, respectively) decrease in ROS levels (
Figure 6A) in HTM cells. The levels of ROS were rescued only partially but not significantly by PDGF treatment (10 ng/ml for 2 h) in cells initially exposed to either DPI or NAC (2 μM or 1 mM respectively for 2 h) as compared to the controls. Both DPI and NAC (2 μM or 1mM, respectively for 2 h) attenuated the effect of Rac1G12V on increase in ROS production in HTM cells (
Figure 6B).
Intracellular ROS generation in TM cells was quantified by flow cytometry using the fluorogenic substrate H2DCFDA. Serum starved HTM cells (3x106 cells/10 cm petri plate) stimulated with PDGF (10 ng/ml for 4 h) (A) or expressing the constitutively active Rac GTPase (B) exhibited significant induction of ROS generation. This response was suppressed when cells were pretreated with either DPI (2 μM for 2 h; inhibitor of NADPH oxidase) or with NAC (1 mM for 2 h; antioxidative agent). Additionally, the Rac GTPase inhibitor EHT1864 or the expression of dominant negative Rac GTPase reduced ROS generation in HTM cells, as compared to untreated controls. Similarly, DPI and NAC both decreased the ROS production in HTM cells significantly compared to untreated cells. (* denotes statistical significance at p < 0.05 based on the mean values of 3 independent experiments). Error bars denote standard error.
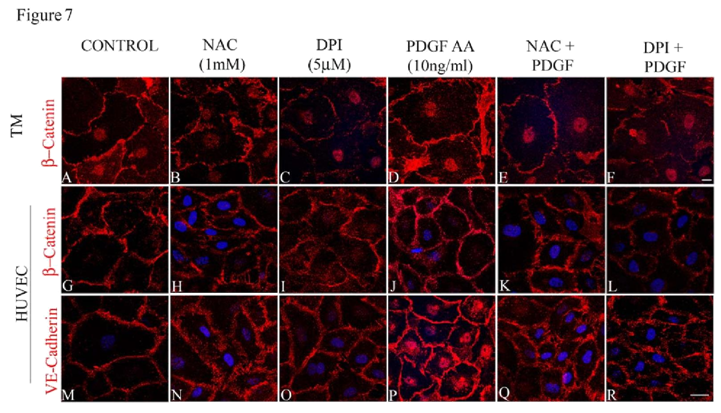
To determine the significance of Rac GTPase-induced ROS production in maintaining the adherens junctions in HTM cells, we scavenged ROS using NAC (1 mM for 2 h) or suppressed the production of ROS with DPI (2 μM for 2 h) after an overnight serum deprivation (1% FBS condition) step. The status of adherens junctions was assessed by immunofluorescence labeling of β-catenin and VEcadherin in NAC/DPI treated HTM and HUVEC cells (Figure 7B, 7C, 7H, 7I, 7N and 7O) relative to control cells (Figure 7A, 7G and 7M), and in the PDGF treated cells (10 ng/ml for 2 h) maintained under serum free conditions (Figure 7D, 7J and 7P). Under these conditions, the adherens junctions staining was not uniform along the cell-cell contacts and resulted in intercellular gaps in NAC and DPI treated cells. The loss of cell-cell junctions as assayed by β-catenin (in both HTM and HUVEC cells) and VE-cadherin (in HUVEC cells) immunostaining was not rescued by PDGF treatment (10 ng/ml for 2 h) in HTM (Figure 7E and 7F) or HUVEC (Figure 7K, 7Q, 7L and 7R) cells pretreated with either NAC or with DPI, demonstrating the importance of ROS in Rac1- induced adherens junctions formation in both HTM and HUVEC cells.
Confluent cultures of HTM or HUVEC cells treated with NAC (1 mM for 2 h) or DPI (2 μM for 2 h) to inhibit ROS generation reveal increased intercellular gaps and decreased staining of β-catenin in both HTM and HUVEC cells and VE-cadherin in HUVEC cells (B, C, H, I, N, O) compared to the control (A, G, M) and PDGF (10 ng/ml for 2 h) treated cells (D, J, P). Treatment with PDGF (10 ng/ml for 2 h) to rescue the NAC/DPI-induced suppression of ROS generation results in partial rescue in terms of stabilization of cell-cell junctions and reduction of intercellular gaps, but not to the level supported by PDGF alone (E, F, K, L, Q, R). Scale bar: 10 μm.
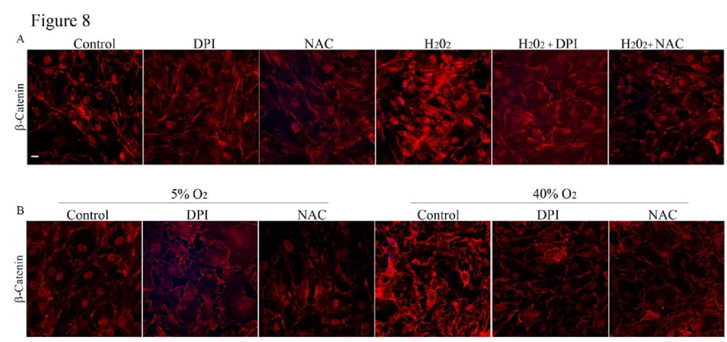
Effects of ROS production induced by H
2O
2 or hyperoxia on adherens junctions and TEER in HTM cells
To further explore the involvement of ROS signaling in maintenance of cell-cell junctions, we assessed the effects of H
2O
2 (250 μM) or 40% oxygen by immunofluorescence labeling of β-catenin in HTM cells. When treated with H
2O
2 (250 μM) for 3 h or under hyperoxia (40% oxygen) for 7 days, the HTM cells displayed strikingly intense β-catenin immunolabeling. But this response of H
2O
2 and hyperoxia was attenuated in the presence DPI or NAC (
Figure 8A and 8B).
A. To further understand the role of ROS and its direct effects on cell-cell junctions, HTM cells in 1% FBS were treated with H2O2 (250 μM) either in the presence or absence of DPI (2 μM) or NAC (1mM). The cells treated with H2O2 (250 μM) alone showed an intense staining of β-catenin compared to the control cells. This response of H2O2 (250 μM) was reduced in the presence of either DPI (2 μM) or NAC (1 mM), which attenuate ROS levels.
B. Similarly to further confirm the participation of ROS in adherens junctions formation in HTM cells, confluent HTM cells cultured in 1% FBS for 24 h were incubatedunder 40% oxygen for 7 days with or without DPI (2 μM) or NAC (1 mM) and assessed for β-catenin immunofluorescence labeling. Simultaneously, another batch of cells was cultured under similar conditions in 5% O2 as control. Immunolabeling for β-catenin in cells cultured under hyperoxic condition for 7 days showed an intense staining compared to the 5% O2 treated control cells. Contrarily, in the presence of DPI (2 μM) or NAC (1 mM), the HTM cells cultured under hyperoxic condition showed no difference in β-catenin immunostaining intensity from the cells maintained under 5% O2 control condition.
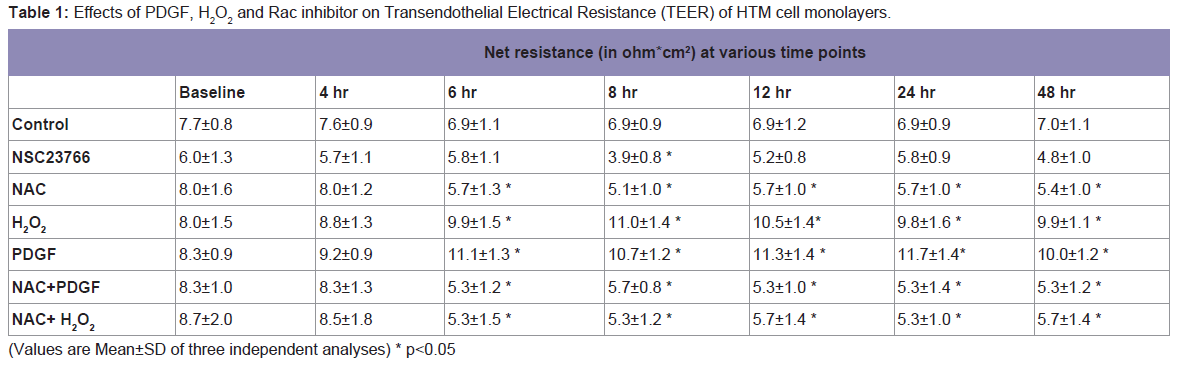
Then to investigate the functional effects of Rac signaling and ROS generation on the TEER, we carried out the measurements of TEER on fully confluent monolayer of HTM cells (
Table 1). As determined by the TEER measurements, the basal net electrical resistance was on an average around 7.9±0.9 ohm.cm
2. Activation of Rac GTPase by PDGF that increases ROS or treatment with H
2O
2 (250 μM) significantly increased the net resistance to 11.1±1.3 and 9.9±1.5 ohm.cm
2, respectively after 6 h of treatment. On the other hand, treatment with Rac inhibitor, NSC23766 (50 μM), showed a significant decrease in the TEER to 3.9±0.8 ohm.cm
2 at 8 h and then the TEER came back to the baseline by 24 h. Treatment with NAC, which quenches ROS, led to a significant decrease in resistance 5.7±1.3 ohm.cm
2 by 6 h and this decrease in resistance was not rescued with the presence of either PDGF or H
2O
2 confirming the significance of ROS in regulation of TM cell TEER (
Table 1). Individual net TEER values are presented in Table 1 as Mean±SD (n=3) and p values are indicated where they are significant.
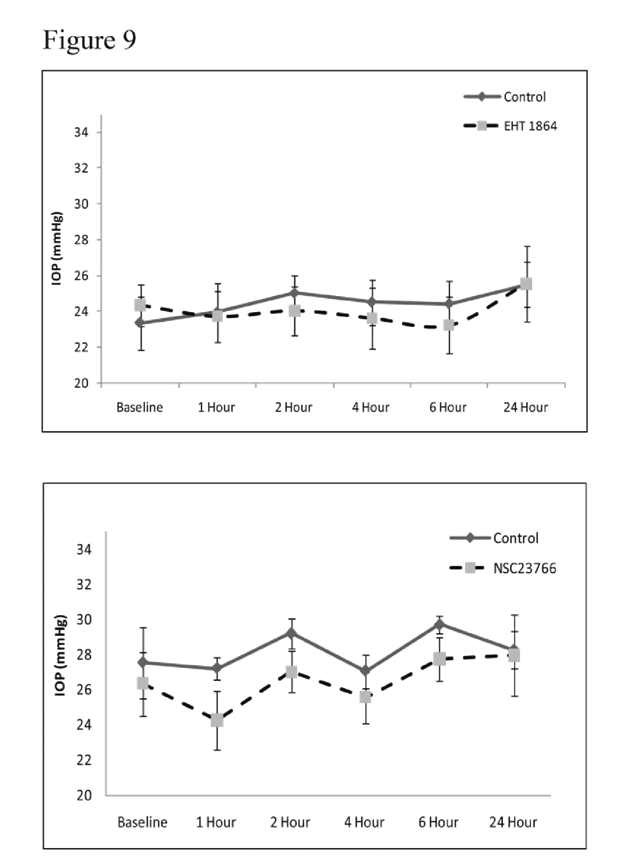
Rac GTPase inhibitors and IOPTo determine the involvement of Rac GTPase signaling in modulation of intraocular pressure (IOP), we tested the effects of Rac GTPase inhibitors on rabbit IOP by topical application of EHT1864 (25 mM) or NSC23766 (50 mM). Maximum concentration of drug that could be dissolved in PBS was used in these experiments. Administration of a single dose (two 30 μl drops) of drug did not cause significant change in IOP relative to controls (vehicle treated, contralateral eye) during the 24 h interval following drug treatment (
Figure 9). The baseline IOP of control eyes (PBS applied) was 24±2 mm Hg for the EHT1864 control group and 26.6±2 mm Hg for the NSC23766 control group. The average decrease in IOP was 2.5±2 mm Hg after 1 h of drug treatment in the case of NSC23766, with the decrease reverting back to the baseline within 2-4 h post-treatment. The decrease in IOP in the drug treated group was not found to be statistically significant from the control group based on Wilcoxon Rank-sum or Student’s t-tests (
Figure 9). Similarly, the EHT 1864 treated group showed no significant drop in IOP compared to the controls (
Figure 9).
To determine the role of Rac GTPase activity in regulation of IOP, the Rac GTPase inhibitors; NSC23766 (50 mM) or EHT1864 (25 mM) were dissolved in PBS, adjusted to pH 7, then applied topically (two 30 μl drops with a one minute delay) to one eye while the other eye was used for the vehicle treatment. The drug was administered only once per animal. Baseline IOP (recorded using a pneumotonometer) in PBS treated control eyes was 24±2 mm Hg (EHT1864 control group) and 26.6±2 mm Hg (NSC23766 control group). IOP changes were monitored for 24 h and there was only a marginal (statistically insignificant, values represent Mean ± SEM, n=6) difference (decrease of 1-2 mm Hg) between the drug treated and control eyes.
To confirm drug penetration through the cornea into the anterior chamber of rabbit eyes described above, drug (NSC23766) concentrations were monitored by LC-MS analysis of AH samples drawn after 1 h of topical application of 50 mM NSC23766. This analysis revealed that an average of ≈50 nM of the drug penetrated into the aqueous humor of rabbit eyes (based on an average of two independent samples). This observation confirms that the drug is capable of penetrating into the AH. The levels of EHT1864 were not determined.
Discussion
Our primary objective of this study was to test the hypothesis that the integrity of adherens junctions in cells of the AH drainage pathway may influence IOP. To test this, here we investigated the regulation of adherens junctions formation in HTM cells in comparison with HUVEC cells by Rac GTPase and assessed the effects of modulators of Rac GTPase activity on IOP in rabbits. This study reveals that a. Rac GTPase and its physiological activator PDGF induce adherens junctions formation in cultured HTM and HUVEC cells in association with changes in actin cytoskeletal reorganization and cell-ECM interactions, b. Rac GTPase activation in HTM cells induces ROS production via NADPH oxidase, c. Suppression of Rac GTPase activity and ROS production in HTM and HUVEC cells impairs adherens junctions formation in both these cell types, d. Hyperoxia, H2O2 and PDGF increase ROS, adherens junctions and transendothelial electrical resistance (TEER) in HTM cells, and finally e. topical application of Rac GTPase inhibitors appears to have a marginal effect on IOP in live rabbits.
Increased resistance to AH drainage through the conventional pathway is the main cause for elevated IOP in primary open glaucoma [
3,
17,18]. Although the precise location of resistance to aqueous drainage within the conventional pathway is not well defined, increased permeability barrier between the inner wall of the SC cells due to increased cell-cell interactions is thought to be one of the possible cellular bases for increased resistance to outflow [
4,5,7,
14]. Although, this possibility has been supported partly by perfusion studies using actomyosin interfering agents, calcium chelators, kinase inhibitors, dexamethasone and growth factors [
4-7,
14,15,
29,
53], the direct role of adherens junctions in IOP homeostasis has not been investigated. To understand the regulation of adherens junctions formation in cells of the AH outflow pathway, which are recognized as the predominant type of cell-cell junctions of endothelial cells [
16,
54,55], we investigated the role of PDGF which is an activator of Rac GTPase signaling [
30,
48]. Stimulation of confluent cultures of the HTM and HUVEC cells with PDGF induced formation of β-cateninbased cell-cell junctions in a Rac GTPase-dependent manner. In contrast, Rac GTPase inhibitors and a dominant negative mutant of Rac GTPase caused a decrease in the basal level of adherens junctions in both HTM and HUVEC cells. PDGF has previously been reported to activate Rac GTPase activity in TM cells [
30]. Moreover, in both HTM and HUVEC cells, expression of constitutively active Rac GTPase induced a robust increase in adherens junctions formation, confirming the direct influence of Rac GTPase signaling on adherens junctions formation in the outflow pathway cells. Similar to the HUVEC cells tested in this study, SC cells which are endothelial in nature, have been reported to develop VE-cadherin based cellcelljunctions [
31]. Therefore, the HUVEC cells used in this study are not identical to SC cells but are expected to serve as a model cell type for SC cells in the context of adherens junctions and permeability characteristics.
To identify the downstream effectors mediating the effects of PDGF and Rac GTPase on adherens junctions in cells of the AH outflow pathway, we measured the levels of ROS which are the product of the Rac activated NADPH oxidase system [
35,
56-58]. Unlike other members of the Rho and Ras family of GTPases, Rac GTPase is one of the components of NADPH oxidase, a multi-component enzyme producing ROS in phagocytic and non-phagocytic cells [
24,
35,
42,
51]. ROS have been shown to play a regulatory role through a redox mechanism [
52,
59] involving for example the tyrosine phosphorylation of various signaling molecules, via targeting the activity of tyrosine phosphatases through cysteine oxidation [
58,60]. Both PDGF and constitutively active Rac GTPase were found to stimulate ROS production significantly in HTM cells. Importantly, inhibition of NADPH oxidase by the flavoprotein inhibitor DPI, or quenching of ROS by the antioxidant NAC in HTM cells, reduced and disrupted the PDGF and constitutively active Rac- induced formation of adherens junctions, confirming that ROS play a critical role in adherens junctions formation in the endothelial cells of the outflow pathway. Additionally, both H
2O
2 and hyperoxia (40% O
2) in HTM cells were found to induce formation of adherens junctions in ROS dependent manner.
In a previous study by Syriani et al. [
30] have reported that perfusion of PDGF increases AH outflow facility in bovine eyes, and topical application of the same compound in rabbits decreases IOP. Although in their study, and based on cell culture observations, the authors implicated Rac GTPase signaling and actin cytoskeletal reorganization as being the main cellular mechanisms accounting for the PDGF induced AH outflow facility and IOP changes, the direct role of Rac GTPase or its downstream effectors has not been confirmed. To address this aspect, we first tested the effects of PDGF and H
2O
2 on transendothelial resistance (TEER) using HTM cells and both these agents were found to increase TEER in ROS dependent manner. We then tested, topically applied Rac GTPase inhibitors on IOP of live rabbits. Intriguingly, the Rac GTPase inhibitors tested in this study did not exhibit significant effect on IOP levels despite confirmation of drug penetration into the anterior chamber of the eye. Although, the effects of Rac inhibitors on IOP were followed only for 24 h in this study, under identical conditions of this study, several other topical agents including Rho kinase inhibitors and autotaxin inhibitor have been reported to influence IOP significantly with one time drug application [
45,
61]. Interestingly, PDGF which was found to increase the adherens junctions formation in HTM and HUVEC cells and TEER in HTM cells, has been reported to lower IOP in rabbits [
30]. On the other hand, Sphingosine-1 phosphate, which has been shown to activate Rac GTPase activity in SC cells has been reported to decrease AH outflow facility in human eyes [
7,
31]. Collectively, these observations indicate that the PDGF induced IOP changes may be independent of Rac GTPase activation and changes in adherens junctions and TEER in the cells of AH outflow pathway.
In an alternative scenario, it is also conceivable that the PDGFinduced decrease in IOP is due to increased Rac GTPase activation leading to ROS production with subsequent destabilization of adherens junctions via ROS-induced phosphorylation of adherens junctional complex proteins, similar to what occurs in microvascular endothelial cells treated with VEGF [
28]. However, in HTM cells, increased ROS production was found to be correlated well with increased TEER. But as a precautionary note we cannot rule out with certainty that the lack of significant changes in IOP in response to inhibitors of Rac GTPase noted in this study was not due to inadequate in vivo levels of drug within the eye.
To unravel a definite role for Rac GTPase in AH outflow facility, future studies are necessary to evaluate the effects of expressing constitutively active and dominant negative mutants of Rac GTPase in the anterior segments of the organ cultured eye on AH outflow facility as well as cell-cell junctions in outflow pathway tissues of different species.
Acknowledgements
The authors thank Pratap Challa, MD., and Philip Lograsso, Ph.D., for their help in collection of aqueous humor from the rabbits and drug analysis in aqueous humor by LC/MS, respectively, and Dr. Toren Finkel, MD. Ph.D., for adenoviral vectors expressing the mutant Rac GTPases. We thank Mr. Christopher Walker for his help with IOP recording and Ms. Kristin M Perkumas for her help in TEER measurements.This work was supported by the grant support of the National Institutes of Health (R01EY018590).
References
- Quigley HA, Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90: 262-267.
- Weinreb RN, Khaw PT (2004) Primary open-angle glaucoma. Lancet 363: 1711-1720.
- Lutjen-Drecoll E (1999) Functional morphology of the trabecular meshwork in primate eyes. Prog Retin Eye Res 18: 91-119.
- Underwood JL, Murphy CG, Chen J, Franse-Carman L, Wood I, et al. (1999) Glucocorticoids regulate transendothelial fluid flow resistance and formation of intercellular junctions. Am J Physiol 277: C330-342.
- Alvarado JA, Yeh RF, Franse-Carman L, Marcellino G, Brownstein MJ (2005) Interactions between endothelia of the trabecular meshwork and of Schlemm’s canal: a new insight into the regulation of aqueous outflow in the eye. Trans Am Ophthalmol Soc 103: 148-162.
- Rao PV, Shimazaki A, Ichikawa M, Franse-Carman L, Alvarado JA, et al. (2005) Effects of novel ethacrynic acid derivatives on human trabecular meshwork cell shape, actin cytoskeletal organization, and transcellular fluid flow. Biol Pharm Bull 28: 2189-2196.
- Sumida GM, Stamer WD (2010) Sphingosine-1-phosphate enhances cortical actomyosin organization in cultured human Schlemm's canal endothelial cell monolayers. Invest Ophthalmol Vis Sci 51: 6633-6638.
- Kumar J, Epstein DL (2011) Rho GTPase-mediated cytoskeletal organization in Schlemm's canal cells play a critical role in the regulation of aqueous humor outflow facility. J Cell Biochem 112: 600-606.
- Clark AF, Brotchie D, Read AT, Hellberg P, English-Wright S, et al. (2005) Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton 60: 83-95.
- Mettu PS, Deng PF, Misra UK, Gawdi G, Epstein DL, et al. (2004) Role of lysophospholipid growth factors in the modulation of aqueous humor outflow facility. Invest Ophthalmol Vis Sci 45: 2263- 2271.
- Zhang M, Maddala R, Rao PV (2008) Novel molecular insights into RhoA GTPase-induced resistance to aqueous humor outflow through the trabecular meshwork. Am J Physiol Cell Physiol 295: C1057-1070.
- Li AF, Tane N, Roy S (2004) Fibronectin overexpression inhibits trabecular meshwork cell monolayer permeability. Mol Vis 10: 750-757.
- Rao PV, Deng P, Sasaki Y, Epstein DL (2005) Regulation of myosin light chain phosphorylation in the trabecular meshwork: role in aqueous humour outflow facility. Exp Eye Res 80: 197-206.
- Kameda T, Inoue T, Inatani M, Fujimoto T, Honjo M, et al. (2012) The effect of Rho-associated protein kinase inhibitor on monkey Schlemm’s canal endothelial cells. Invest Ophthalmol Vis Sci 53: 3092-3103.
- Rao PV, Deng PF, Kumar J, Epstein DL (2001) Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Invest Ophthalmol Vis Sci 42: 1029-1037.
- Komarova Y, Malik AB (2010) Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72: 463-493.
- Fautsch MP, Johnson DH (2006) Aqueous humor outflow: what do we know? Where will it lead us? Invest Ophthalmol Vis Sci 47: 4181-4187.
- Johnson M (2006) ‘What controls aqueous humour outflow resistance?’. Exp Eye Res 82: 545-557.
- Tian B, Gabelt BT, Geiger B, Kaufman PL (2009) The role of the actomyosin system in regulating trabecular fluid outflow. Exp Eye Res 88: 713-717.
- Alvarado JA, Betanzos A, Franse-Carman L, Chen J, Gonzalez-Mariscal L (2004) Endothelia of Schlemm’s canal and trabecular meshwork: distinct molecular, functional, and anatomic features. Am J Physiol Cell Physiol 286: C621-634.
- O’Brien ET, Perkins SL, Roberts BC, Epstein DL (1996) Dexamethasone inhibits trabecular cell retraction. Exp Eye Res 62: 675-688.
- Heimark RL, Kaochar S, Stamer WD (2002) Human Schlemm’s canal cells express the endothelial adherens proteins, VE-cadherin and PECAM-1. Curr Eye Res 25: 299-308.
- Sumpio BE, Riley JT, Dardik A (2002) Cells in focus: endothelial cell. Int J Biochem Cell Biol 34: 1508-1512.
- Wojciak-Stothard B, Ridley AJ (2002) Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39: 187-199.
- Lum H, Malik AB (1994) Regulation of vascular endothelial barrier function. Am J Physiol 267: L223-241.
- Dejana E, Orsenigo F, Lampugnani MG (2008) The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121: 2115-2122.
- Dudek SM, Garcia JG (2001) Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487-1500.
- Monaghan-Benson E, Burridge K (2009) The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem 284: 25602-25611.
- Burke AG, Zhou W, O’Brien ET, Roberts BC, Stamer WD (2004) Effect of hydrostatic pressure gradients and Na2EDTA on permeability of human Schlemm’s canal cell monolayers. Curr Eye Res 28: 391-398.
- Syriani E, Cuesto G, Abad E, Pelaez T, Gual A, et al. (2009) Effects of platelet-derived growth factor on aqueous humor dynamics. Invest Ophthalmol Vis Sci 50: 3833-3839.
- Stamer WD, Read AT, Sumida GM, Ethier CR (2009) Sphingosine-1-phosphate effects on the inner wall of Schlemm’s canal and outflow facility in perfused human eyes. Exp Eye Res 89: 980-988.
- Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ (2001) Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 114: 1343-1355.
- Spindler V, Schlegel N, Waschke J (2010) Role of GTPases in control of microvascular permeability. Cardiovasc Res 87: 243-253.
- Vandenbroucke E, Mehta D, Minshall R, Malik AB (2008) Regulation of endothelial junctional permeability. Ann N Y Acad Sci 1123: 134-145.
- Frey RS, Ushio-Fukai M, Malik AB (2009) NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal 11: 791-810.
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101: 7618-7623.
- Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ (2008) Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol 439: 111-129.
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, et al. (2007) Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem 282: 35666-35678.
- Pattabiraman PP, Rao PV (2010) Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am J Physiol Cell Physiol 298: C749-763.
- Pracyk JB, Tanaka K, Hegland DD, Kim KS, Sethi R, et al. (1998) A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest 102: 929-937.
- Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, et al. (1996) rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol 16: 7115-7121.
- Rao PV, Maddala R, John F, Zigler JS Jr. (2004) Expression of nonphagocytic NADPH oxidase system in the ocular lens. Mol Vis 10: 112-121.
- Liton PB, Lin Y, Luna C, Li G, Gonzalez P, et al. (2008) Cultured porcine trabecular meshwork cells display altered lysosomal function when subjected to chronic oxidative stress. Invest Ophthalmol Vis Sci 49: 3961-3969.
- Perkumas KM, Stamer WD (2012) Protein markers and differentiation in culture for Schlemm’s canal endothelial cells. Exp Eye Res 96: 82-87.
- Iyer P, Lalane R 3rd, Morris C, Challa P, Vann R, et al. (2012) Autotaxin-lysophosphatidic acid axis is a novel molecular target for lowering intraocular pressure. PLoS One 7: e2627.
- Feng Y, Yin Y, Weiser A, Griffin E, Cameron MD, et al. (2008) Discovery of substituted 4-(pyrazol-4-yl)- phenylbenzodioxane-2-carboxamides as potent and highly selective Rho kinase (ROCK-II) inhibitors. J Med Chem 51: 6642-6645.
- Hawkins PT, Eguinoa A, Qiu RG, Stokoe D, Cooke FT, et al. (1995) PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol 5: 393-403.
- Chiariello M, Marinissen MJ, Gutkind JS (2001) Regulation of c-myc expression by PDGF through Rho GTPases. Nat Cell Biol 3: 580-586.
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10: 778-790.
- Ganitkevich V, Hasse V, Pfitzer G (2002) Ca2+-dependent and Ca2+-independent regulation of smooth muscle contraction. J Muscle Res Cell Motil 23: 47-52.
- Bokoch GM, Diebold BA (2002) Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 100: 2692-2696.
- Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. (1996) Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J 318: 379-382.
- Clark AF, Wilson K, de Kater AW, Allingham RR, McCartney MD (1995) Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest Ophthalmol Vis Sci 36: 478-489.
- Franke WW (2009) Discovering the molecular components of intercellular junctions--a historical view. Cold Spring Harb Perspect Biol 1: a003061.
- Dejana E (1997) Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest 100: S7-S10.
- Bokoch GM (1994) Regulation of the human neutrophil NADPH oxidase by the Rac GTP-binding proteins. Curr Opin Cell Biol 6: 212-218.
- Irani K, Goldschmidt-Clermont PJ (1998) Ras, superoxide and signal transduction. Biochem Pharmacol 55: 1339-1346.
- Finkel T (2006) Intracellular redox regulation by the family of small GTPases. Antioxid Redox Signal 8: 1857-1863.
- Pracyk JB, Tanaka K, Hegland DD, Kim KS, Sethi R, et al. (1998) A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy. J Clin Invest 102: 929-937.
- Chiarugi P, Buricchi F (2007) Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid Redox Signal 9: 1-24.
- Honjo M, Tanihara H, Inatani M, Kido N, Sawamura T, et al. (2001) Effects of rho-associated protein kinase inhibitor Y-27632 on intraocular pressure and outflow facility. Invest Ophthalmol Vis Sci 42: 137-144.